





研究交流
異所性肝臓の構築
大橋一夫
東京女子医科大学先端生命医科学研究所
- はじめに
- 肝ティッシュエンジニアリングの経緯
- 肝ティッシュエンジニアリングのアプローチ
- 腎被膜下肝組織作製法
- 皮下肝組織作製法
- 異所作製肝組織の機能
- マウス内ヒト肝組織作製
- 異所作製肝組織の3次元化
- 作製肝組織の増殖による3次元化法
- シート工学による皮下肝組織の3次元化
-
肝細胞の大量確保にむけて
- マウス内肝細胞増殖法
- 自己細胞を用いての治療の可能性
- さいごに
- 文献
1. はじめに
肝臓疾患における未来型医療の展開を模索する中で、我々の研究グループは生体第二の肝臓組織を作製する肝ティッシュエンジニアリングの開発に従事している。肝ティッシュエンジニアリングの研究は緒についたばかりの新しい分野であるが、異所的部位(肝臓以外の部位)において作製肝組織が機能すれば、持続的な機能補助をもたらすことが可能となる1,2)。つまり、その機能が小さいものであっても、多くの肝疾患で病態の進行を防ぎ、QOL向上をもたらすことが期待される。成熟肝細胞を利用して組織を作製することが基盤となるが、肝細胞の多機能性を維持することが重要である。我々は特に、作製肝組織の機能維持の長期化と、作製肝組織の3次元化を目標に研究をすすめてきた。本稿では、肝ティッシュエンジニアリングの経過につき振り返り、今後の展望について述べたい。
2. 肝ティッシュエンジニアリングの経緯
肝細胞を生体内の異所的部位に生着させる開発は、世界で約30年の研究歴を有する。作製部位は、大網内、腸間膜内、皮下脂肪内、肺動脈内、肺実質内あるいは膵臓内等の報告がなされている。コラーゲンをコートしたマイクロビーズや、ポリ乳酸を基盤とした生分解性スカフォールドと肝細胞の混合体を生体内に移植する手法が主にとられてきた3)。異所肝細胞へ門脈血流に多く含まれる増殖因子を供給することを目的として、門脈―下大静脈シャントを作製して増殖因子を大循環系に流す試みもなされていた。しかしながら、多くの報告において、異所肝細胞の観察期間が1週―数週と限られた期間であり、作製肝組織を安定させる手法は確立されていない状況にあった。そのため、中・長期的な組織維持に必用な要素が解明されておらず、組織機能についての情報も乏しい。また、肝臓の魅力的特徴である再生増殖現象について、自己肝臓と異所肝組織が如何なる連携を成し得るかという課題についても、解明がまたれていた。
3. 肝ティッシュエンジニアリングのアプローチ
3-1. 腎被膜下肝組織作製法
腎被膜下部位は、豊富な腎実質血流に近接することと、腹膜からの栄養供給を受けることからも、肝細胞のような代謝活性の高い細胞の生着には有利な部位である。また、免疫反応から遠い部位―いわゆるimmunoprivilegedな部位とされており3)、他家肝細胞利用を視野に入れた場合にも重要な部位である。また、腎臓外への細胞拡散がないために、局所での細胞検索が容易に行い得る。これらのことから、腎被膜下部位において、分離肝細胞を長期生着させることを目指した。
同系マウスの実験系において、分離肝細胞を浮遊状態で腎被膜下に注入するのみでは、これまでの諸家らの報告と同じく、多くの細胞が移植後1週間で死に至る。腎被膜下における細胞死は、酸素供給不足ではなく、細胞外基質との相互作用不足による接着不良がもたらすものと考えられたため、IV型コラーゲンやラミニンに富むEHSマトリックスとともに肝細胞を移植する試みを行った5-9)。その結果、肝細胞相互の接着が促進し、腎被膜下において1―2層の肝細胞索構造を有する肝組織を形成する5-9)。この肝組織はマウスの生涯を通じ(400日以上)維持されるものである(図1A,B)が、安定して組織量が維持されることは、散発的に発生する肝細胞死による欠損分を、隣接肝細胞が緩徐に分裂することで組織全体の機能量を補填することに起因している6)。
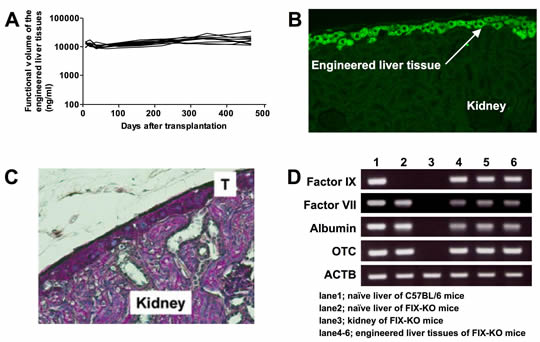
図1 異所性肝組織(腎被膜下)の長期安定性
A-C、human alpha1-antitrypsin(hA1AT)トランスジェニックマウス肝細胞をEHSマトリックスとともに野生型マウスの腎被膜下に移植し、肝組織を作製した。A、腎被膜下作製肝組織の機能的組織量の推移。レシピエント血中hA1AT濃度にて組織量を検索したところ、組織作製後450日(マウス生涯の約2/3に相当)の観察期間を通じて作製肝組織は安定して推移する。B、作製肝組織のhA1AT免疫蛍光染色。作製肝組織は、1-2層の肝細胞索からなる薄層組織である。C、PAS染色。D、血友病B(FIX-KO)モデルマウスの腎被膜下に野生型マウス肝細胞を用いて作製した肝組織のmRNA発現。FIX-KOマウスをレシピエント、同系野生型マウスを肝細胞ドナーとしている。レーザーマイクロダイセクション法にて作製肝組織、自己肝、腎臓のmRNA発現量をRT-PCRで検索した。(一部は文献6, 20を改変し、引用)
3-2. 皮下肝組織作製法
新規治療法を開発するにあたり、治療の侵襲度をより小さくすることも重要な目標である。「皮下」は最もアプローチが容易で、局所麻酔下での治療が行えることから、ティッシュエンジニアリングの標的部位として魅力的部位である1,2,10)。注射感覚での肝疾患再生医療が可能となれば、真の夢治療である11)。また、経過観察中に、腫瘤形成等の万一予期せぬ事態が発生した際にも、最も対処し易い部位であることから、将来的な幹細胞由来細胞や遺伝子改変細胞を利用した治療にも有用な部位と考えている。
「皮下」の問題点は、肝細胞の生着が他の部位と比較して著しく不良なことである。ハーバード大のMooneyら12)は、スカフォールドを改良し、HGF/VEGF/EGFをカクテルに徐放するシステムを搭載しているが、皮下での肝細胞生着期間は1週間にとどまっている。我々の研究過程においても、肝細胞への細胞外マトリックスの供給あるいは、増殖因子を供給する工夫を行ったものの、細胞生着向上への大きな寄与は得られなかった5,6) 。そこで、皮下の乏血管性が最大の問題点と考え(図2A)、組織作製予定局所にあらかじめ血管ネットワークを構築し、次いで肝細胞を移植する2段階法を開発した(図2B)13-15)。aFGF徐放マイクロスフェアーズ6)やbFGF徐放デバイス16,17)を局所に挿入し(図3A,B)、血管ネットワークを誘導するものである(図3C)。この2段階法により、長期間(100日以上)安定して肝細胞は生着し、肝組織を形成した。作製肝組織は肝臓と同レベルで凝固第VIII/IX因子の遺伝子発現を行う(肝細胞当りの評価)16)ことから、血友病や遺伝性酵素欠損症の補充療法に有用なアプローチとなる2, 11, 18)。このような皮下への組織作製手技は、新生児患者にも低侵襲で行い得る手技であることから、次世代再生治療の開発に重要な要素技術と考えている。
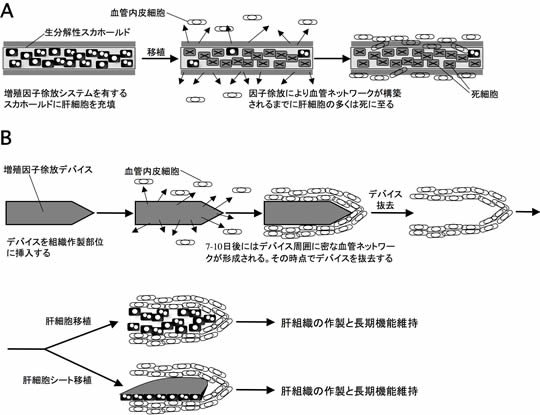
図2 皮下への肝ティッシュエンジニアリングのアプローチ
A、生分解性スカホールドを用いた従来型アプローチ。肝細胞をスカホールド内に充填して移植。細胞増殖因子を徐放するシステムを搭載したスカホールド等が開発されてきているが、肝細胞への即時的な酸素/栄養供給が十分でないため、長期的な細胞生着が得られていない。B、血管ネットワークをあらかじめ誘導した後に肝細胞を移植する2段階的アプローチ。血管内皮細胞増殖因子を徐放するデバイスをあらかじめ挿入することにより血管内皮細胞に富む皮下プラットフォームを作製する。この段階で細胞または肝細胞シート移植(図7参照)を行うと、長期間機能する肝組織作製が可能となる。
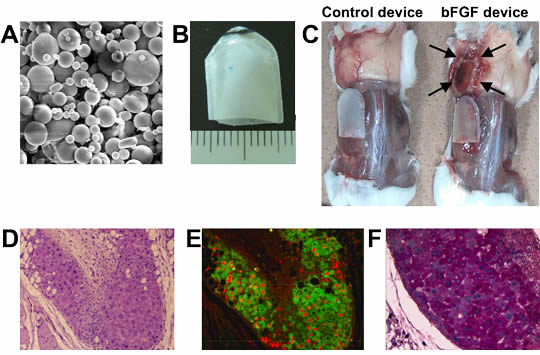
図3 皮下への血管ネットワーク作製と作製肝組織
A、acidic FGFを充填したマイクロスフェアーズ。皮下局所に注入し、血管ネットワークを構築する。B、basic FGF充填メッシュバッグデバイス。ポリエチレンテレフタレートのメッシュ膜をバック状にし、ポリビニルアルコールで膜を補強している。内腔にアガロースに溶解したbasic FGFを充填する。C、basic FGFデバイス皮下挿入による血管ネットワーク構築。コントロールデバイス(左)と比較してbasic FGFデバイス周囲には非常に密な血管ネットワークが形成されている(右、矢印)(挿入後10日目)。D-F、血管ネットワークを構築した皮下空間に、EHSマトリックスとともに肝細胞を移植して作製した皮下肝組織。自己肝に部分切除を加えることで皮下作製肝組織も増殖する。D、H&E染色。E、hA1AT(緑)BrdU(赤)免疫染色。F、PAS染色。(一部は文献6, 16を改変し、引用)
3-3. 異所作製肝組織の機能
機能的な組織を作製することが、肝ティッシュエンジニアリングの最大の目標である。上述の手法により作製し得た腎被膜下および皮下の肝組織は、自己肝臓とほぼ同等の機能を発揮する(組織量当り)。各種血液凝固因子、アルブミン、アンチトリプシン、OTC等の産生および糖新生において機能を確認している(図1C,D)5,6,16,19,20)。さらに、薬剤をレシピエントマウスに投与した際には、作製肝組織も薬剤を取り込み、対応する薬物代謝酵素を誘導する16,17,20)。薬物以外にも、本来肝臓が取り込むべき様々な化合物を取り込む。また、これら化合物を取り込む速度は、自己肝臓と遜色ないものである(未発表データ)。これらのことは、血流を介する十分な物質交換をサポートする構造、つまり、類洞内皮細胞を含めた肝臓特異的構造を有していることを示している。これら化合物の代謝後の排泄は、毛細胆管へと排泄するが、異所性作製肝組織には排泄管としての胆管を持たないため、おそらくは周囲血流へ拡散し、大循環系を経たのち、自己肝臓で胆汁排泄しているものと推測している。
異所生着肝細胞が、血液凝固因子の産生機構を有していることは興味深いものである(図1D)。従来、肝細胞培養下では培養早期に血液凝固因子産生が低下または消失することが課題であった。しかしながら、腎臓被膜下および皮下の肝ティッシュエンジニアリング過程においてはこの機能低下は発生しない11,16,19-21)。血液凝固因子発現において培養下と異所生着下の、発現調節機構の異なりは不明であり、今後の解明に期待したい。肝ティッシュエンジニアリングにおいて、作製肝組織が凝固因子を供給し、血友病病態に治療効果をもたらすことを、血友病A(FVIII-KO)および血友病B(FIX-KO)マウスモデルで確認している16,19)。
3-4. マウス内ヒト肝組織作製
肝ティッシュエンジニアリングの手法を用いて、免疫不全マウス内にヒト肝組織を作製することも可能である1,5,29)。EHSマトリックスは、ヒト肝細胞のマウス異所性生着にも効果を発揮する7)。我々は、EHSマトリックスと共に肝細胞を移植する手法に加え、ヒトcMet特異的な作動性抗体を作製し、レシピエントマウスに投与した5,22)。この2手法の組み合わせにより、マウス内異所ヒト肝組織を100日以上安定させ、ヒト肝組織を有するキメラマウスを作出することに成功した。ヒト肝組織キメラマウスにB型肝炎ウイルスと注入すると、マウス内ヒト肝組織がB型肝炎ウイルスの感染・複製・放出を担い、ウイルス血症を呈する(図4A,B)5)。また、このB型ウイルス血症マウス内のヒト肝組織は、D型肝炎ウイルスにも感染し、D型肝炎血症を呈した(図4C,D)5)。この結果は、肝ティッシュエンジニアリングによるヒト肝組織の作製が、肝炎ウイルス感染の小動物モデル開発への道を開くことを世界で初めて示し得たものである。
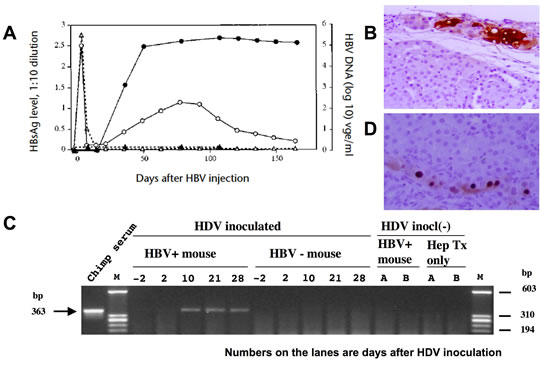
図4 ヒト肝組織キメラマウス作製による肝炎ウイルス感染モデル
NOD/SCIDマウスの腎被膜下にヒト肝細胞をEHSマトリックスと共に移植後、ヒトcMET作動性抗体を投与することで、安定して機能するヒト肝組織を作製し得る。A、このヒト肝組織キメラマウスにHBV患者血清を注入することによる、B型肝炎ウイルス血症の発生。ヒト肝組織キメラマウス(●○)およびヒト肝組織を持たないNOD/SCIDマウス(▲△)血液中のHBs抗原レベル(○△)とHBVゲノムレベル(●▲)。B、B型肝炎ウイルス血症を呈するヒト肝組織キメラマウスの腎被膜下ヒト肝組織のHBs抗原染色。C、B型肝炎ウイルス血症となったヒト肝組織キメラマウスへD型肝炎ウイルスを注入することによるD型肝炎ウイルス血症の発生。D、D型肝炎ウイルス血症を呈するヒト肝組織キメラマウスの腎被膜下ヒト肝組織のHD抗原染色。(一部は文献3を改変し、引用)
4. 異所作製肝組織の3次元化
さて、上述の手法により長期組織維持が可能となっているものの、作製肝組織は細胞索が1-2列の(2次元的な)小肝組織である。より多くの病態に対応できる治療手段として確立することを目指し、組織3次元化を図り肝組織の機能量を増す手法の開発に着手している。
4-1. 作製肝組織の増殖による3次元化法
肝臓は機能量が低下した際には、すみやかで活発な再生増殖を行い、定常機能レベルの獲得後にはすみやかに再生を停止する。この再生増殖は、肝臓研究分野を魅了してきた。しかし、異所的に作製・存在する肝組織の再生増殖に関する情報は極めて少ない。我々は、腎被膜下および皮下に肝組織を作製後70日目に自己肝の2/3切除を行い、増殖力を評価した。その結果、図5,6に示すごとく、自己残肝の再生に伴い、作製肝組織も約2.6倍へと増殖し、3-4個の肝細胞索の3次元組織へと成長することが明らかとなった(図5B,C)6)。肝切除後BrdUを2週間徐放し細胞周期が進行した肝細胞数を評価したところ、自己肝臓では93%、作製肝組織で91%とほぼ同じ増殖活性を発揮することが示されている(図6B,E,F)6)。皮下作製肝組織でも同様である(図3D,E)6, 17)。これら作製肝組織の活発な再生増殖は、組織内の特殊な肝細胞が増殖を担っているのではなく、作製組織内のほぼ全ての肝細胞が増殖する。この組織全体が協調して成される増殖は、組織工学的アプローチにおける成熟肝細胞の有用性を示唆するものである。
さらに次にあげる重要な点も明らかとなった。①異所肝組織の再生増殖は、自己肝臓の再生停止に伴って停止する6,9)。②再生増殖を行った異所作製肝組織は、疲弊することなく長期間維持される9)。③再生増殖は繰り返しが可能で、より組織幅の厚い3次元組織へと発育する(図5D)9)。④薬剤誘導によるdirect hyperplasia型の再生増殖も、自己肝と同レベルで発揮する(図6I,J)6)。⑤肝組織を作製し、長期間経過した後であっても、再生増殖力は衰えない(図6A,B,G,H)20)。これらの異所性肝組織の再生現象は、良好な組織血流により液性因子が十分に供給されていることと、組織内において肝細胞―非実質細胞間のクロストークが構築されていることを示している。多くの肝疾患において、肝障害に伴う肝再生刺激が生体内で発生していることから、初期作製組織が薄層組織であっても、経緯とともに3次元化組織へと変化していく可能性を提唱したい。
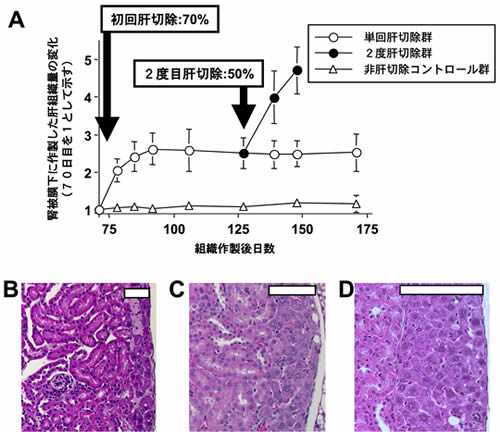
図5 腎被膜下製肝組織の再生力
A、腎被膜下作製肝組織の機能的組織量の推移。EHSマトリックスとともに分離肝細胞を腎被膜下に移植することで、長期間安定する肝組織が作製される。作製肝組織は、1-2層の肝細胞層から成る薄層肝組織である(B)。このマウスの自己肝に2/3肝部分切除を行うことにより肝再生刺激を誘導すると、自己肝の再生に連動して作製肝組織も約2.6倍に増殖し、3-4層から成る肝組織へと成長する(C)。残肝に再切除(50%肝切除)を行うと、再度の再生増殖により5-6層の肝組織へと成長する(D)。(B-DはH&E染色。barは肝組織幅を示す)(一部は文献6, 9を改変し、引用)
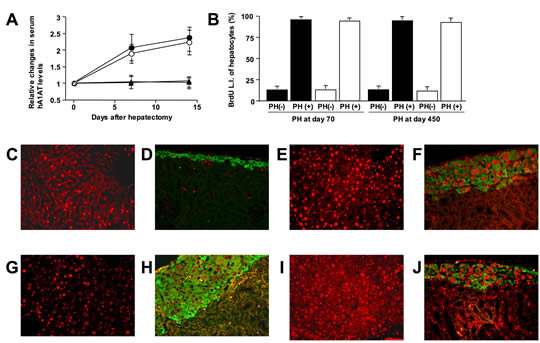
図6 異所性肝組織(腎被膜下)の再生増殖活性
マウス腎被膜下にhA1ATトランスジェニックマウス肝細胞を用いて肝組織を作製し、70日後または450日後に自己肝の2/3切除を行うことで、再生刺激を誘導した。BrdUを肝切除後から14日間徐放し、細胞周期が進行した肝細胞を検索した。A、レシピエント血中hA1AT濃度にて評価した作製肝組織量の推移。●▲; 70日後実施群、○△; 450日後実施群。●○;2/3肝切除施行、▲△;sham手術(非肝切除)施行。B、BrdU肝細胞標識率。■;自己肝臓、□;作製肝組織。PH;2/3肝切除。C-J; hA1AT(緑) BrdU(赤)蛍光免疫染色。C,D;非肝切除コントロール、E,F;組織作製後70日目に2/3肝切除施行し14日後の摘出標本、G,H;組織作製後450日目に2/3肝切除施行し14日後の摘出標本、I,J;組織作製後70日目に薬剤誘導型direct hyperplasia肝再生を誘導し14日後の摘出標本。C,E,G,I;自己肝臓、D,F,H,J;作製肝組織 。(一部は文献6, 8, 9, 20を改変し、引用)
4-2.シート工学による皮下肝組織の3次元化
皮下における3次元肝組織構築法を開発するにあたり、我々の着目した点は、肝細胞相互の機能接着と肝細胞―血管内皮細胞の接着を制御することであった。ばらばらの肝細胞を局所に注入するだけでは、細胞相互が接着できるかどうかは細胞側の動態にたよるものであり、一定の生着効率を確保することが困難である(図2)。そこで、温度応答性インテリジェント培養皿を用いて肝細胞を培養することにより、細胞相互の接着を保ったまま、シート状組織として回収し、肝細胞シートを貼布することによる肝ティッシュエンジニアリングを開発した(図2)。
温度応答性インテリジェント培養皿は、ポリN-イソプロピルアクリルアミドをナノメートルレベルで均一に共有固定化した培養皿である。このポリマーの下限臨界温度(32℃)以上では、ポリマーは疎水性であることから通常培養皿と同じく接着培養を行い得る(図7A)23)。一方、培養温度を下限臨界温度以下に下げると、ポリマーが親水性に変化し、培養皿接着面から細胞を持ち上げ、細胞間接着に影響を与えることなく細胞がシート状として回収できる。つまり、温度応答性インテリジェント培養皿を用いてコンフルエント状態となった初代培養肝細胞(図7A)は、一時的な培養温度低下(15-30分)のみにより、肝細胞シートとして回収できる(図7B,C)。この肝細胞シートの特徴は、単に細胞が接着したシート状組織ではなく、肝細胞が培養経過中に細胞相互間に形成した毛細胆管、デスモゾーム、タイトジャンクション等の機能的細胞間結合がそのまま保たれた組織であることにある(図7D)。さらに、細胞が産生する細胞外マトリックスをシート底面に保持しており、高次機能の発揮に適した細胞外微小環境を有している17)。
この肝細胞シートを、あらかじめ血管ネットワークを構築した皮下空間に貼布移植することで、生体内での即時的な肝細胞―血管内皮細胞の接合をなし得る(図7E-H)。つまり、肝細胞が相互に結合した2次元組織の両側を血管内皮層がサンドウィッチする3次元組織体が皮下に作製される。この手法により、半永久的に機能が維持される肝組織が皮下に作製される。また、肝細胞シートを複数枚積層することにより、組織幅のより厚い皮下肝組織も作製可能となっている17)。
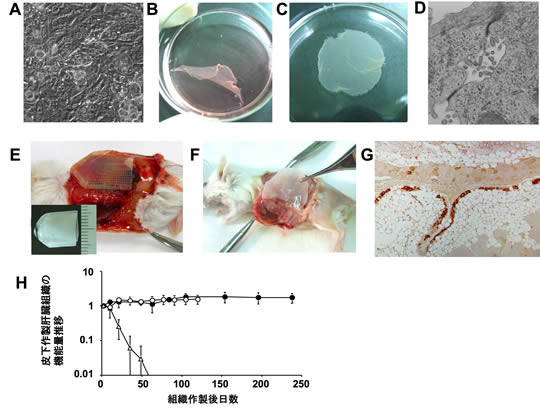
図7 肝細胞シートを用いた皮下への肝ティッシュエンジニアリング
温度応答性高分子(PIPAAm)を固定化した温度応答性インテリジェント培養皿にて肝細胞培養を数日間行うとコンフルエント培養状態となる(A)。培養温度を一時的に20度に下げることで、肝細胞は自然に培養皿から剥離し(B)、肝細胞をシート状組織として回収できる(C)。このシート状組織は、培養中に肝細胞相互が形成する毛細胆管等の機能的細胞接合を保った組織である(D)。E、組織工学を目的とした血管ネットワークを誘導した皮下プラットフォーム(図3参照)。F、basic FGFデバイスを皮下に挿入後7-10日程度経過した後に、デバイスを摘出して皮下プラットフォームを開き、肝細胞シートを貼布移植する(F、紙状膜をキャリアーとしてシートを移植)。G、皮下に作製される2次元的肝組織。H、皮下作製肝組織の組織機能量の推移。肝細胞シートを血管ネットワークを誘導した皮下プラットフォームへ貼布した群(○●)と、通常皮下へ貼布した群(△)。(一部は文献17を改変し、引用)
5. 肝細胞の大量確保にむけて
ヒト肝細胞は脳死提供肝臓や手術摘出肝組織片から分離して用いられる1)。分離肝細胞は、通常培養下では増殖しない。現在のところ、門脈血流を経由して病態肝臓へ移植する肝細胞移植が世界の数施設で行われ、一部の症例では肝臓移植に匹敵する治療効果も報告されている。肝ティッシュエンジニアリングが今後発展し、肝細胞治療が全体としてレベルアップするためには、十分量の肝細胞数を確保することが重要である。我々は、特に肝細胞の血液凝固因子産生能に着目して18,23)、マウス体内において凝固因子の発現力を維持しつつヒト肝細胞を増殖させる手法を確立した25)。
5-1. マウス内肝細胞増殖法
uPA/SCIDマウスという肝障害を持続的に発生するマウスの肝臓内に少数のヒト肝細胞を移植するものである(uPA/SCIDマウスについては肝細胞研究会ホームページにて詳細な紹介がなされているので参照いただきたいhttp://hepato.umin.jp/kouryu03.html)。マウス内移植ヒト細胞は活発かつ持続的に増殖し、約10週後にはマウス肝臓の大部分を置換する(図8)25)。レシピエントuPA/SCIDマウスの血液中では、ヒト正常レベルのヒト凝固第IX因子濃度が検出され、凝固生理活性を伴っている。一方、通常培養下での肝細胞は、培養早期に凝固因子発現力を消失する。つまり、凝固因子発現―翻訳・翻訳後修飾―分泌という凝固因子産生の全過程を維持した肝細胞を大量に供給する現存唯一の手法と考えている26)。
血液凝固因子を産生する細胞を幹細胞の分化誘導により確保する試みも、重要な課題である。ヒト第VIII遺伝子ユニットを挿入したマウスES細胞で胚様体を形成し、内胚葉系に分化させることで、高効率な第VIII因子分泌が可能となっており27)、さらなる発展が期待されている。
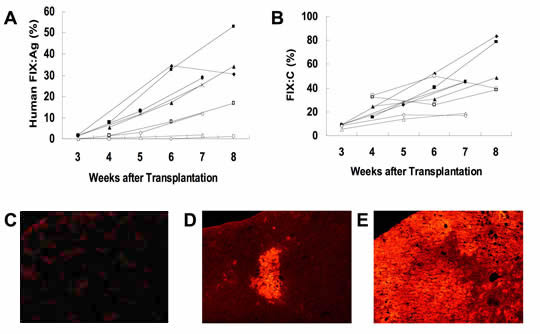
図8 ヒト肝細胞の大量増幅システム
uPA/SCIDマウスの肝臓へヒト肝細胞を移植し、移植後の増殖過程をマウス血中のヒト凝固第IX因子濃度(A)、ヒト凝固第IX因子活性(B)、マウス肝臓のヒト第IX因子染色(C-E)で評価。非移植コントロール(C)、ヒト肝細胞の増殖が開始した直後の低置換肝臓(D)、増殖が進みマウス肝臓をほぼ置換した肝臓(E)。少数のヒト肝細胞がuPA/SCIDマウス肝臓内で第IX因子産生能を維持しつつ継続的に増殖し、マウス肝をほぼ置換するに至る。A, Bの各ラインは9匹uPA/SCIDレシピエント個別のデータを示す。(一部は文献25を改変し、引用)
5-2. 自己細胞を用いての治療の可能性
上述の治療戦略は、凝固因子発現が正常な他家(アロ)肝細胞を用いるものであり、免疫抑制法の付加が必須である。一方、自己肝細胞を大量に確保する方法が確立できれば、免疫抑制法を必用としない細胞治療が成立する。我々は、遺伝性肝疾患の肝細胞がuPA/SCIDマウスシステムで大量増殖し得るかの検討を目的に、肝細胞からの凝固第IX因子産生が欠損することに起因する血友病Bを一例として取り上げ、血友病Bマウス肝細胞によるuPA/SCIDマウス置換実験を行った。uPA/SCIDマウス肝臓は、血友病Bマウス肝細胞による置換が徐々に進行し、その結果、図9に示すごとく、凝固第IX因子活性が低下した。最終的には99%以上の置換により凝固第IX因子活性は測定下限以下(正常の0.5%以下)となり、完全な血友病B病態へと変化した。この実験結果は、uPA/SCIDマウスシステムにおいて、遺伝性疾患の肝細胞も大量増殖が可能なことを示している。この増殖肝細胞を回収し、種々な遺伝子治療ベクターを用いて、遺伝子修飾を施すことが可能となれば28)、自己肝細胞による肝ティッシュエンジニアリングという新しい遺伝性肝疾患治療の開発に結びつくものと期待している。
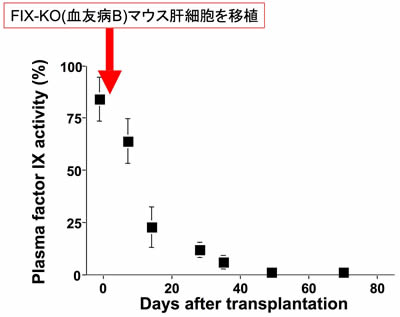
図9 血友病B肝細胞によるuPA/SCIDマウス肝臓置換
血友病Bモデル(FIX-KO)マウスの肝細胞をuPA/SCIDマウス肝臓に移植。uPA/SCID肝臓内で増殖置換がすすむにつれ、レシピエントマウスの凝固第IX因子活性は著明に低下し、6週後には完全な血友病B病態となる。uPA/SCIDマウス肝臓の99.5%以上が血友病B肝細胞で置換されることが遺伝子解析にて判明している。
6. さいごに
肝細胞を利用した再生医療は、多くの肝疾患に対する新しい治療展開として期待できるものである。第二の肝組織を作製するアプローチは、ロマンに満ちた未来型医療と考えている。肝細胞と非実質細胞との相互構築、3次元組織化、遺伝子修飾ならびに作製組織に対する免疫反応制御等が今後の臨床応用へむけての開発事項である。また、肝臓病態と作製肝組織の長期的なバランスを知ることも重要である。肝細胞は、高次機能を有する有用な細胞である。しかし、肝臓/組織片提供者の年齢や背景のばらつき、分離肝細胞の純度・機能・構造にかなりのバリエーションが存在する。このバリエーションを理解しつつ、多種多様な肝病態へ工夫をこらして適応させることを目指していきたいと考えている。
謝辞
本編記載の研究成果の一部は、京都大学再生医科学研究所(岩田博夫教授)、広島大学理学部(吉里勝利教授)、奈良県立医科大学消化器・総合外科(中島祥介教授)、同小児科(吉岡章教授)との共同研究のもとに遂行したものであり、ご協力いただいた教室員の先生方に深く御礼申し上げます。
7. 参考文献
- Ohashi K, Park F, Kay MA. Hepatocyte transplantation: Clinical and experimental application. J Mol Med 79: 617-630, 2001.
- Ohashi K. Liver tissue engineering: The future of liver therapeutics. Hepatology Res 38:S76-87, 2008.
- Demetrious AA, Levenson SM, Novikoff PM, et al. Survival, organization, and function of microcarrier-attached hepatocytes transplanted in rats. Proc Natl Acad Sci USA 83:7475–7479, 1986.
- Horne PH, Lunsford KE, Walker JP, et al. Recipient immune repertoire and engraftment site influence the immune pathway effecting acute hepatocellular allograft rejection. Cell Transplantation 17: 829-844, 2008.
- Ohashi K, Marion PL, Nakai H, et al. Sustained survival of human hepatocytes in mice: A model of in vivo infection with human hepatitis B and hepatitis delta viruses. Nature Med 6:327-331, 2000.
- Ohashi K, Waugh JM, Dake MD, et al. Liver tissue engineering at extra-hepatic sites in mice as a potential new therapy for genetic liver diseases. Hepatology 41:132-140, 2005.
- Ohashi K, Kay MA. Extracellular matrix component co-transplantation prolongs survival of heterotopically transplanted human hepatocytes in mice. Transplant Proc 36: 2469-2470, 2004.
- Kuge H, Ohashi K, Yokoyama T, et al. Genetic modification of hepatocytes towards hepatocyte transplantation and liver tissue engineering. Cell Transpl 15:1-12, 2006.
- Ohashi K, Kay MA, Yokoyama T, et al. Stability and repeat regeneration potential of the engineered liver tissues under the kidney capsule in mice. Cell Transpl 14:50-59, 2005.
- Fox IJ, Schafer DF, Yannam GR. Finding a home for cell transplantation: Location, location, location. Am J Transpl 6: 5-6, 2006.
- 大橋一夫. 今後の展望―肝ティッシュエンジニアリング 白幡聡編:みんなに役立つ血友病 医薬ジャーナル社2009, 309-317.
- Smith MK, Riddle KW, Mooney DJ. Delivery of hepatotrophic factors fails to enhance longer-term survival of subcutaneously transplanted hepatocytes. Tissue Eng 12: 235-244, 2006.
- 大橋一夫、中島祥介. 肝細胞シートを用いた肝組織工学の新展開 医学のあゆみ 220:581-586, 2007.
- 大橋一夫. 肝組織工学―組織3次元化技術開発による新展開 日本移植学会雑誌 43: 312-139, 2008.
- Sole K. New device aids bioengineering hepatic tissue. Nature Clin Prac Gastro Hepatol 3: 183, 2006.
- Yokoyama T, Ohashi K, Kuge H, et al. In vivo engineering of metabolically active hepatic tissues in a neovascularized subcutaneous cavity. Am J Transpl 6:50-59, 2006.
- Ohashi K, Yokoyama T, Yamato M, et al. Engineering functional two- and three-dimensional liver systems in vivo using hepatic tissue sheets. Nature Med 13:880-885, 2007.
- Pipe SW, High KA, Ohashi K, et al. Progress in the molecular biology of the inherited bleeding disorders. Haemophilia 14(S3):130-137, 2008.
- Ohashi K, Tatsumi K, Utoh R, et al. Liver tissue engineering under the kidney capsule site provides therapeutic effects to hemophilia B mice. (manuscript in submission).
- Ohashi K, Koyama F, Tatsumi K, et al. Functional life-long maintenance of engineered liver tissue in mice following transplantation under the kidney capsule. (manuscript in submission).
- 大橋一夫、中島祥介. 肝細胞移植の現状と展望―血友病の次世代治療の確立にむけて 日本血栓止血学会雑誌 15:541-546, 2004.
- Ohashi K, Meuse L, Schwall R, et al. cMet activation allows persistent engraftment of ectopically transplanted xenogenic human hepatocytes in mice. Transplant Proc 38: 587-588, 2001.
- Yang J, Yamato M, Shimizu T, et al. Reconstruction of functional tissues with cell sheet engineering. Biomaterials 28: 5033-5043, 2007.
- Tatsumi K, Ohashi K, Shima M, et al. Therapeutic effects of hepatocyte transplantation on hemophilia B. Transplantation 86:167-170, 2008.
- Tatsumi K, Ohashi K, Kataoka M, et al. Successful in vivo propagation of factor IX-producing hepatocytes in mice: Potential for cell-based therapy in haemophilia B. Thromb Haemost 99:883-891, 2008.
- Ofosu F. Propagating factor IX-producing hepatocytes for haemophilia B therapy. Thromb Haemost 99:799-800, 2008.
- Kasuda S, Kubo A, Sakurai Y, et al. Establishment of embryonic stem cells secreting human factor VIII for cell-based treatment of hemophilia A. J Thromb Haemost in press, 2008.
- Park F, Ohashi K, Chiu W, et al. Efficient lentiviral transduction of liver requires cell cycling in vivo. Nat Genet 24:49-52, 2000.

