





研究交流
NASH進展における脂質代謝ホメオスタシスの破綻とその意義
中川 勇人
三重大学大学院医学系研究科消化器内科学
はじめに
非アルコール性脂肪性肝疾患(nonalcoholic fatty liver disease; NAFLD)は肝細胞への脂肪沈着を特徴とし、中でも非アルコール性脂肪肝炎(nonalcoholic steatohepatitis; NASH)は、炎症・線維化を伴い肝硬変や肝癌に進展し得る疾患である。肝細胞への脂肪蓄積はNASHのfirst hitとして重要と考えられ、現在脂質生合成に関わる酵素を標的とした治療薬の開発が盛んに行われている。しかし我々は、①強力かつ広範囲にわたる脂質生合成阻害はかえって肝病態を悪化させうること、②進行したNASHの一病態である“burned-out NASH”においても脂質生合成機能が低下しており、病態悪化に寄与している可能性があること、を明らかにした。そこで本稿では同研究結果を中心に、NASH病態進展における脂質代謝異常の意義について概説する。
SREBPを中心とした脂質生合成とNAFLD/NASH
アセチルCoAを出発点として、Acetyl-CoA carboxylase(ACC)、Fatty acid synthase(FASN)、sterol-CoA desaturase 1(SCD1)などの酵素反応を介して脂肪酸を合成する過程を、de novo脂肪酸合成と呼ぶ。NAFLD患者ではde novo脂肪酸合成が亢進しており、健常者では肝組織中脂質の80%が肝外由来脂質でありde novo合成されたものは5%しかないが、NAFLD患者ではde novo合成脂質の割合が26%にも達すると報告されている1。そのため上記酵素を標的とした薬剤の臨床試験が現在進行中である2。しかしこれまでのところ、これらの薬剤単剤では十分なNASH改善効果が得られているとは言い難い。
我々はこれまで、脂質生合成に関わる様々な酵素の発現を正に制御するマスターレギュレーター転写因子、Sterol regulatory element-binding protein (SREBP)に着目して研究を行ってきた。SREBPは脂肪酸合成やコレステロール合成に関わる様々な酵素の発現を制御する転写因子で、脂質生合成の司令塔の役割を担っている(図1)。前述のACC 、FASN、 SCD1といった脂質生合成酵素はいずれもSREBPによって制御されていることから、SREBP阻害によってより効率的にNASHを治療できるのではないかと考え、実際にSREBPを阻害する新しい化合物の開発にも取り組んできた 3。加えて、SREBPを介したde novo脂肪酸合成は肝細胞癌においても高頻度に活性化しており、肝癌に対する治療標的としても有望な可能性がある4。しかしこれまで、SREBPの阻害がNASH進展・発癌を予防するという、明確な実験的エビデンスは存在しなかった。
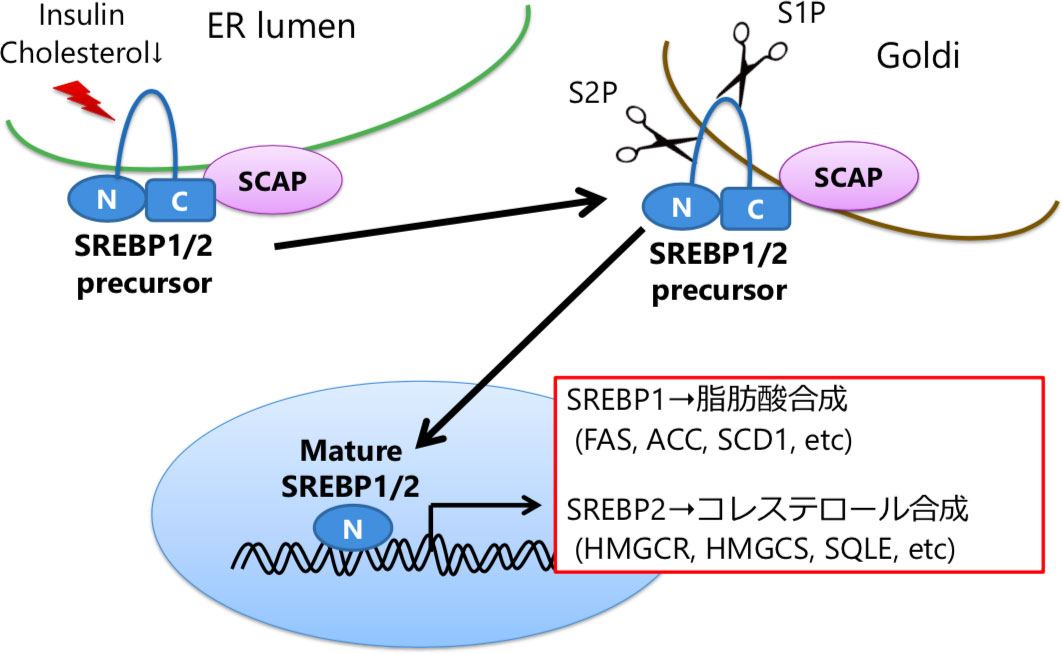
図1. SREBPの活性化機構
SREBPは膜結合型の前駆体として小胞体膜に存在し、活性化の際にはエスコート蛋白であるSCAP によってゴルジ装置に輸送される。次いでSREBPはプロテアーゼによって切断を受け、N末端部分が活性型として核内に移行し、標的遺伝子の転写を活性化する。SREBPはSREBP-1a、SREBP-1cそしてSREBP-2という3つのアイソフォームからなり、SREBP-1cは主に脂肪酸合成、SREBP-2は主にコレステロール合成を制御し、さらにSREBP-1aは両者の制御に関わっている。
NASH肝癌マウスモデルにおけるSREBP阻害効果
これらの背景から我々は、NASH進展・発癌におけるSREBP阻害の長期効果について、マウスモデルを用いて検証した5。NASH肝癌モデルとして、SREBP活性化を伴ってNASHから肝細胞癌を発症する肝臓特異的PTEN欠損マウス(Alb-Cre;PTENflox/flox→PTEN KOマウスと表記)を用い6、同マウスにさらにSREBP活性化に必須の分子SREBP cleavage-activated protein(SCAP)を欠損させたマウスを交配させ、肝臓特異的PTEN/SCAPダブルノックアウトマウスを作成した(Alb-Cre;PTENflox/flox;SCAPflox/flox →PS-DKOマウスと表記)。図1に示すように、SREBPは活性化の際にエスコートタンパクSCAPにより小胞体からゴルジ体に輸送される必要があり、SCAPが欠損するとSREBP活性化がほぼ完全に阻害される7。結果、PS-DKOマウスでは予想通り肝細胞の脂肪滴形成が著明に抑制されたが、肝細胞障害・炎症はむしろ悪化し、5か月齢で肝硬変、さらに7か月齢で多発肝癌を発症するという全く予想に反する結果となった(図2)。一方同週齢のPTEN KOマウスは脂肪肝にはなるものの炎症・線維化は軽度で、SCAP KOマウスはほぼ正常であった。またコリン欠乏高脂肪食を用いたNASH肝癌モデルにおいてSCAPを欠損させても同様の結果が得られた。さらに、PS-DKOマウスの肝臓に活性型SREBPを導入すると細胞死や炎症、発癌が著明に改善した。これらの結果から、NASHにおいて脂質生合成を強力に阻害することは、むしろ肝病態を悪化させうる可能性が示唆された。
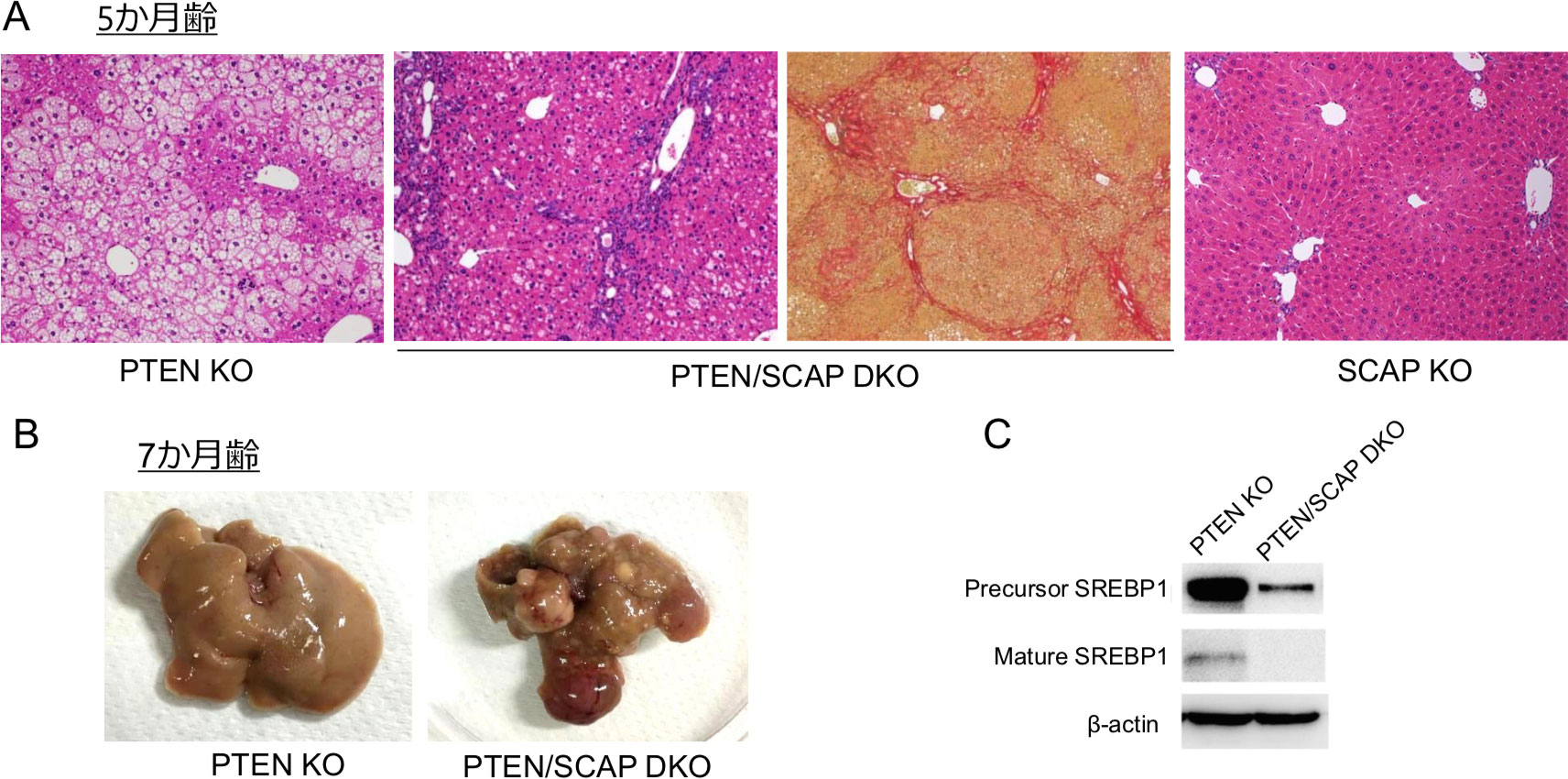
図2. NASH肝癌マウスモデルにおけるSREBP阻害効果
A. 各マウスの5か月齢での肝臓病理像。PTEN-SCAP DKOの右画像のみSirius Red染色。ほかはH&E染色。
B. 7か月齢のPTEN KOマウスとPTEN/SCAP DKOマウスの肝臓肉眼像。
C. 5週齢のPTEN KOマウスとPTEN/SCAP DKOマウスの肝臓から抽出したタンパクを用いたWestern blotting。PTEN/SCAP DKOマウスではほぼ完全にSREBPの活性化が抑制されている。
(Kawamura S, et al. 20225より改変引用)
強力なSREBP阻害は小胞体膜のリン脂質組成変化を介して小胞体ストレスを惹起する
各genotypeのマウス肝組織を用いてトランスクリプトーム解析を行ったところ、PS-DKOマウスの肝臓では小胞体ストレスシグナル経路の顕著な活性化を認め、電子顕微鏡でも小胞体の障害が観察された。PS-DKOマウスの肝臓に小胞体ストレス軽減作用を持つ分子シャペロンGRP78を導入したところ、有意に肝障害が改善したことから、同マウスの肝障害には小胞体ストレスが重要な役割を果たしていることがわかった。
脂質生合成阻害による小胞体ストレス発症機序を明らかにするため、網羅的リピドミクス解析を行った。するとPS-DKOマウスでは、ホスファチジルコリン(phosphatidylcholine ; PC)やホスファチジルエタノールアミンといったリン脂質に組み込まれている脂肪酸の組成が大きく変化していた(図3A)。リン脂質は細胞膜や細胞内小器官などの生体膜の構成成分として重要で、親水性の頭部構造と疎水性の2本の脂肪酸鎖が結合した構造をしており、その組み合わせによって1000種類を超えるリン脂質分子種が存在する。興味深いことにPS-DKOマウスでは、C20:4などの多価不飽和脂肪酸(poly-unsaturated fatty acid; PUFA)を含むPCが減少する一方で、C16:0など飽和脂肪酸のみからなるPCが増加するという不均衡が生じていた(図3B)。リン脂質の多様性は生体膜の構造・機能維持に必須であり、特に不飽和脂肪酸を含むリン脂質は細胞膜や小胞体膜の流動性維持に重要と考えられている8。小胞体は、タンパク質の合成、折り畳み、輸送などの役割を担っているが、これらの機能を維持するには小胞体膜の流動性が必要であり、膜流動性が低下すると異常なタンパクが小胞体に蓄積し小胞体ストレスの原因となる。そこでPS-DKOマウス由来初代肝細胞に、小胞体膜に取り込まれる特性を持つリポソームを用いてPUFAを含むPCを導入したところ、小胞体ストレスが有意に減弱した。さらにPS-DKOマウスにPCカクテルを補充することによって、小胞体ストレス・肝障害が有意に改善したことから、脂質生合成阻害は小胞体膜におけるリン脂質組成異常を介して小胞体ストレス・肝障害を惹起すると考えられた(図3C)。またSREBP機能不全は、de novo脂肪酸合成の低下に加え、LXRの活性低下を介してPUFAをPCへ組み込む酵素であるLPCAT3の発現を低下させ、リン脂質の組成変化を惹起していることも明らかとなった。
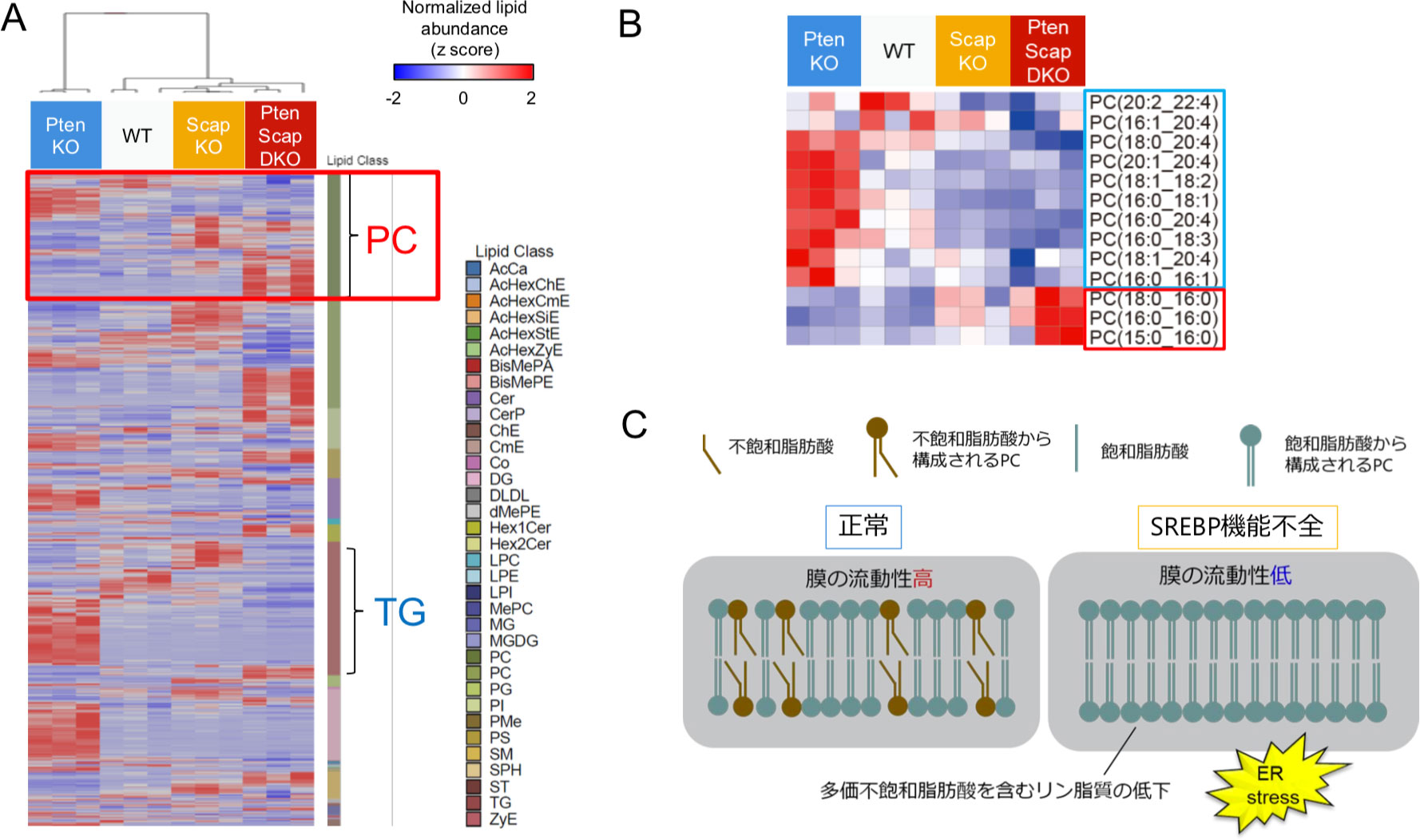
図3. SREBP機能不全はリン脂質の脂肪酸組成変化を介して小胞体ストレスを惹起する
A. 5週齢の各マウスの肝臓を用いたLC-MSによる網羅的脂質解析。PC: phosphatidylcholine, TG: triacylglycerol。
B. Aのヒートマップのうち、PCの部分を拡大。
C. 多価不飽和脂肪酸を含むPCの低下は、小胞体膜の流動性低下によって小胞体ストレスを惹起する。
(Kawamura S, et al. 20225より改変引用)
進行したNASHの一病態“burned-out NASH”ではSCAP/SREBP経路の活性が低下している
肝細胞への脂肪蓄積はNAFLDのfirst hitとして重要と考えられる一方で、肝線維化が進行したNASHでは、肝組織中の脂肪沈着はむしろ減少・消失し、“burned-out NASH”と呼ばれる状態となり、かえって肝硬変や肝発癌のリスクが高まることが知られている。しかしながら、burned-outという現象が起きる原因およびその病態における意義はほとんどわかっていない。そこでヨーロッパ9および自施設のNASH肝生検RNA-seqデータを用いてヒトNAFLDの発現プロファイルを網羅的に解析したところ、興味深いことに肝線維化進行に伴ってSCAP-SREBP-LXR-LPCAT3 axisの活性低下が生じていた。よって同経路の活性低下はburned-out現象の一因となっているとともに、進行NASHのさらなる病態進展に寄与している可能性が示唆された(図4A)。
おわりに
最近ヒトNASHにおいても病態進展とともにリン脂質組成が変化することが報告されており10, 11、本研究結果と合わせて、リン脂質の補充やリン脂質への脂肪酸組み込み異常の是正が、進行NASHの治療法の一つとなることが期待される。また現在NASHに対して脂質生合成経路を標的とした薬剤の開発が盛んに行われているが、今回の研究結果から、過剰かつ広範な脂質生合成阻害はかえって病態を悪化させる可能性も示唆された。このことは今後の薬剤開発において重要な示唆を与えると思われ、おそらくSREBP活性を適切なレベルに保つことが重要と考えられる。加えて、初期のNASHとburned-out NASHでは、病態における脂質代謝経路の働きが大きく変化しており(図4B)、将来的には個々のステージに応じて、より個別化された治療戦略が必要になると考えられる。
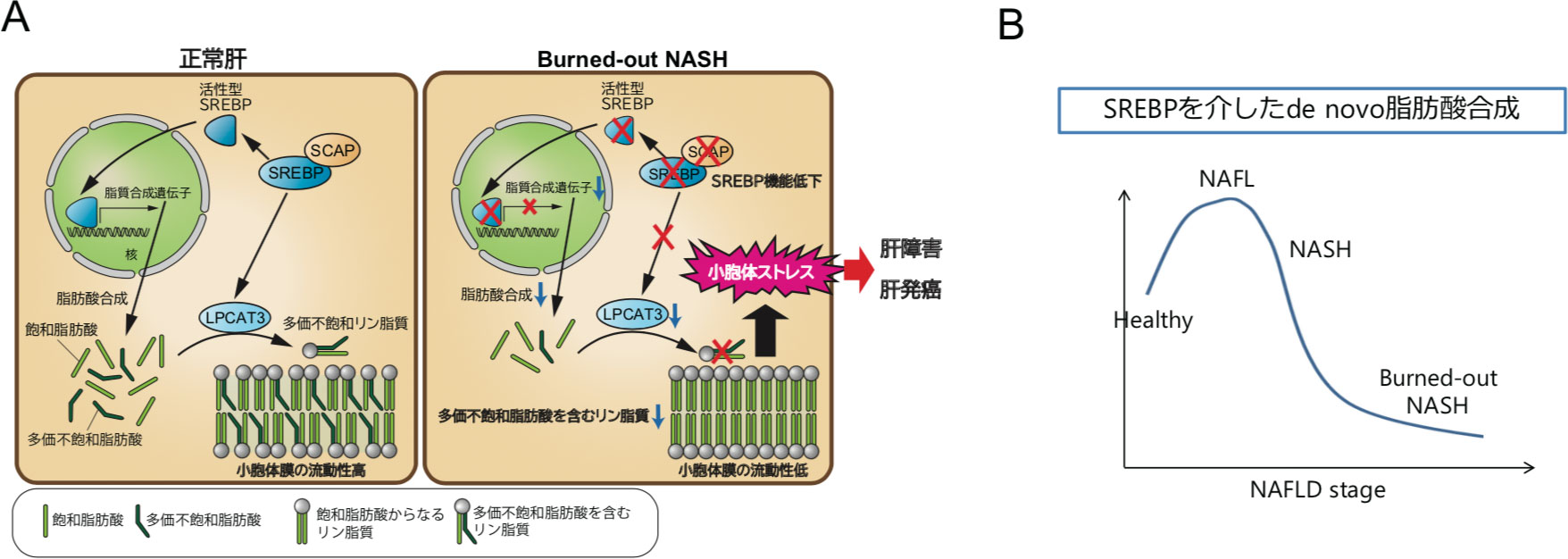
図4. SREBP機能低下から肝障害が生じる仕組み
A. 研究結果のまとめ。Burned-out NASHでは、SCAPの発現低下を伴ってSREBPの活性が低下している。SREBP機能障害は、de novo脂肪酸合成低下とLPCAT3発現低下を介して生体膜リン脂質に組み込まれる脂肪酸の組成を変化させる。その結果小胞体膜の流動性が低下し、小胞体ストレスを引き起こす。
B. NAFLDの病態進展とSREBPを介した脂肪酸合成活性との関係。
文献
- Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343-51.
- Nakagawa H. Treatment for nonalcoholic steatohepatitis -current status and future prospects of pharmacotherapy. Nihon Shokakibyo Gakkai Zasshi 2021;118:829-839.
- Kawagoe F, Mendoza A, Hayata Y, et al. Discovery of a Vitamin D Receptor-Silent Vitamin D Derivative That Impairs Sterol Regulatory Element-Binding Protein In Vivo. J Med Chem 2021;64:5689-5709.
- Nakagawa H, Hayata Y, Kawamura S, et al. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers (Basel) 2018;10.
- Kawamura S, Matsushita Y, Kurosaki S, et al. Inhibiting SCAP/SREBP exacerbates liver injury and carcinogenesis in murine nonalcoholic steatohepatitis. J Clin Invest 2022;132.
- Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest 2004;113:1774-83.
- Matsuda M, Korn BS, Hammer RE, et al. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev 2001;15:1206-16.
- Rong X, Albert CJ, Hong C, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab 2013;18:685-97.
- Govaere O, Cockell S, Tiniakos D, et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci Transl Med 2020;12.
- Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007;46:1081-90.
- Hall Z, Bond NJ, Ashmore T, et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017;65:1165-1180.

