





研究交流
肝細胞における低酸素・再酸素化傷害
- 光プローブを用いたプログラム細胞死(ネクロトーシス・アポトーシス)の解析 -
芳賀 早苗a、尾崎 倫孝a,b
a 北海道大学大学院 保健科学研究院 生体応答制御医学分野
b 北海道大学大学院 保健科学研究院 生体分子・機能イメージング部門
概要
肝傷害は、肝炎、肝切除後あるいは肝腫瘍に対する塞栓療法後など様々な病態下で観察される。肝傷害は、主としてネクローシスあるいはプログラム細胞死(アポトーシスなど)の結果として発生する。今回、肝細胞死による肝傷害を研究するために、すでに開発済みのアポトーシス検出用光プローブ(casapse3活性)と新たにデザイン・作製したネクロトーシス検出用光プローブ(receptor-interacting protein [RIP]1/RIP3結合)をもちいて、低酸素後(虚血後)の肝傷害におけるそれぞれのプログラム細胞死の役割を比較検討した。
ネクロトーシスを感知するプローブとして、ネクロトーシス誘導の主経路に位置するRIP1/RIP3の結合(二量体化)を利用し、RIP1/RIP3が結合することにより分割したルシフェラーゼ断片(N末片およびC末片)が再構成され活性型ルシフェラーゼ(full length)形成する光プローブをデザイン・作製した。
マウス肝細胞株TIB73において、TNF-α/cycloheximide(T/C)刺激だけではRIP1/3結合を誘導しなかったが、加えてcaspase阻害剤zVAD-fmk(zVAD)によりcaspase活性を抑制することでRIP1/3結合を誘導した。このRIP1/3結合は、RIP1のアロステリック阻害剤であるnecrostatin-1(Nec-1)によって阻害された。また、T/C/zVADによる細胞生存率の低下はNec-1投与により改善した。これらの事実は、T/Cは、それ自体ではcaspase3経路を活性化してアポトーシスを誘導するが、caspaseが阻害され(あるいは利用不能となり)アポトーシスが誘導されない時には、それに代わってネクロトーシスが誘導される可能性を示している。
Fasリガンド(FasL)により誘導される肝細胞死は、zVAD投与によって部分的に阻害されたのみであったため、FasL刺激による細胞死にはアポトーシス以外のプログラム細胞死が関与する可能性が示唆された。FasL刺激もT/C刺激と同様に、caspaseが不活性化された時にのみにRIP1/3結合を誘導した。興味深いことに、FasLにより誘導されるRIP1/3結合は、抗酸化剤であるTroloxあるいはN-acetyl
cysteine(NAC)により有意に抑制されたため、FasLにより誘導されるネクロトーシス経路には酸化ストレスの関与が示唆された。H2O2はその刺激のみでRIP1/3結合を誘導したが、zVADによりcaspase活性を阻害してもRIP1/3結合は増強しなかった。
低酸素・再酸素化(H/R)刺激はRIP1/3結合と細胞死を誘導したが、細胞死はNec-1および抗酸化剤によって抑制された。H2O2刺激と同様に、H/RによるRIP1/3の結合誘導にはcaspase阻害を必要としなかったため、caspase非依存的な機構の存在が示唆された。酸化ストレスはネクロトーシス誘導に関与するが、従来のcaspase依存性経路とは異なるnon-canonicalな経路を介している可能性がある。
はじめに
肝傷害は肝臓内の様々な病的状態を反映し、肝細胞をはじめとした種々の構成細胞の傷害を伴う1-8。肝疾患, 肝臓に対する治療(癌に対する化学療法、肝臓の外科的切除、肝移植による虚血/再灌流[ischemia/reperfusion [I/R]など)においても、ネクローシス、アポトーシスといった細胞死が生じることで肝が傷害される。したがって、肝細胞死の観点から肝傷害・肝細胞死の原因・プロセスを理解することは、臨床的にも重要であると考えられる。
いくつかのタイプの細胞死は細胞内シグナル伝達により制御され、かつレドックス依存的に発生すると言われている6-10。アポトーシスによる細胞死は、急性および慢性肝炎、あるいは虚血後の肝傷害6,11など、様々な肝の病的状態で発生することがよく知られている。例えば、Fasリガンド(FasL)は、主にcaspaseおよびレドックス依存的にアポトーシスを誘導するが7、これは術後肝傷害、B/C型肝炎、アルコール性肝炎12-15など多くの病態に関与している。Fas抗原(CD95)は、肝細胞(非腫瘍・腫瘍に関わらず)に対して恒常的に発現するため16、FasL/Fas(CD95)による細胞死は癌および非癌状態における様々な病的細胞死に関与するものと考えられる。
近年、TNFαなどのアポトーシス誘導タンパク質が特定の条件下でネクローシスを誘発することが確認されており17、ネクロトーシス(necroptosis)と呼ばれている。TNFαは細胞死と細胞生存の2つのシグナル伝達経路を活性化する。細胞死のシグナルは、それぞれアポトーシス誘導またはネクローシス誘導シグナル伝達経路に分かれる。TNFαが細胞のTNFα受容体に結合すると、TRADD(TNF
receptor-associated death domain)、TRAF 2(TNF-receptor-associated factor 2)、TRAF 5、RIPK 1(receptor interacting protein kinase 1 [RIP1])などから成るComplex Iを形成する。これらのタンパク質は通常ポリユビキチン化されるが、A20(TNFIP3)、cezanne(OTUD7B)およびUSP21(ubiquitin specific
peptidase), RIPK1などにより脱ユビキチン化されると、Complex IIに移行する。Complex IIは、RIPK1、RIPK3、Fas-associated death domain(FADD)、TRADDおよびcasapse8等からなる。Complex IIのcaspase8が活性化されると、RIPK1およびRIPK3は活性を失い、活性化caspase8は下流のcaspase(caspase3、6、および7)を活性化し、caspase依存性細胞死であるアポトーシスを誘導する。この際、細胞がcaspase阻害剤(zVAD)で処理されていると、細胞はネクローシス様細胞死“ネクロトーシス(Necroptosis)”を引き起こす。RIPK1 の活性化はこのプロセスに重要な役割を果たすことがわかっている。RIPK1が活性化され自己リン酸化し、さらにRIPK3をリン酸化(活性化)し、二量体化することが報告されている。RIPK1/RIPK3の二量体化(活性化)からネクロトーシス誘導までの分子メカニズムに関しては様々な報告があるが、その詳細な機序の多くは未だ明らかになっていない。
近年、腎の虚血・再灌流傷害、心筋梗塞および急性膵炎といった病態にネクロトーシスが関与している可能性が示唆されている17-21。しかし、肝臓・肝細胞のネクロトーシスは十分に研究されておらず、肝の生理病態との関連性については未だ明らかではない。最近、肝におけるTNF誘導性ネクロトーシスのメカニズムが報告されているが、それは多くの炎症性肝疾患に対して重要な機構と考えられる22-24。また、ネクロトーシスは腫瘍形成時にしばしば抑制されていることが明らかになっているため、薬剤による癌治療25の標的となることが期待されている。
ネクロトーシス誘導に関して最も深く研究されている機序の1つは、TNFα/TNF受容体(TNFR)などのリガンド/受容体刺激によって誘導される経路である。TNFα/TNFRは、ネクロトーシス誘導に不可欠なプロセスであるRIP1およびRIP3の物理的相互作用(RIP1/3結合)を誘導する。もうひとつはFasL/Fas(CD95)による誘導機序であり、これも非アポトーシス性の細胞死を誘導することが知られており、ネクロトーシスの概念に一致している25。また、上述のごとくネクロトーシスはcaspase活性が抑制された状況下で(アポトーシスが起こりにくい条件下で)誘導されるため、アポトーシスが十分に進行しない場合に備えた「バックアップ機構」とも考えられている。
我々は、RIP1とRIP3の物理的相互作用を感知してネクロトーシスを検出する新しい光学プローブをデザイン・作製した。これまでに作製しているアポトーシス検出のための光プローブ(caspase3活性化光プローブ)とこのネクロトーシス検出のための光プローブ(RIP1/3結合)を用いて、肝細胞死のダイナミックな解析を試みた。これらの手法は、種々の細胞死の相互関係、病態形成における役割を研究するために有用なツールとなると考えている。
MEF細胞において、RIP1/3結合は、TNF-α/ cycloheximide または Sodium Azide(NaN3)投与によって増加した
生細胞のネクロトーシスをモニターするために、RIP1とRIP3の物理的相互作用を特異的に検出する光プローブを図1Aに示す。この光プローブを遺伝子導入したマウス線維芽細胞MEFを用いて、TNF-α/cycloheximide(T/C)あるいはNaN3(sodium azide)を投与した。T/CおよびNaN3は、各々リガンドおよび非リガンド刺激によりプログラム細胞死を誘導するが、少なくとも部分的にはcaspase依存性アポトーシスを介している33, 34。MEF細胞において、T/Cは投与後4時間までに急激にRIP1/3結合の増加を誘導し、その後の24時間で細胞死を誘導した(図1B)。NaN3は、投与後RIP1/3結合とMEF細胞死を誘導したが、その後細胞死の増加に伴い速やかに低下した(図1C)。
ネクロトーシス検出用の光プローブは、T/CおよびNaN3処置MEFで細胞死を引き起こすRIP1/3結合(すなわちネクロトーシス)を評価することが可能であると考えられた。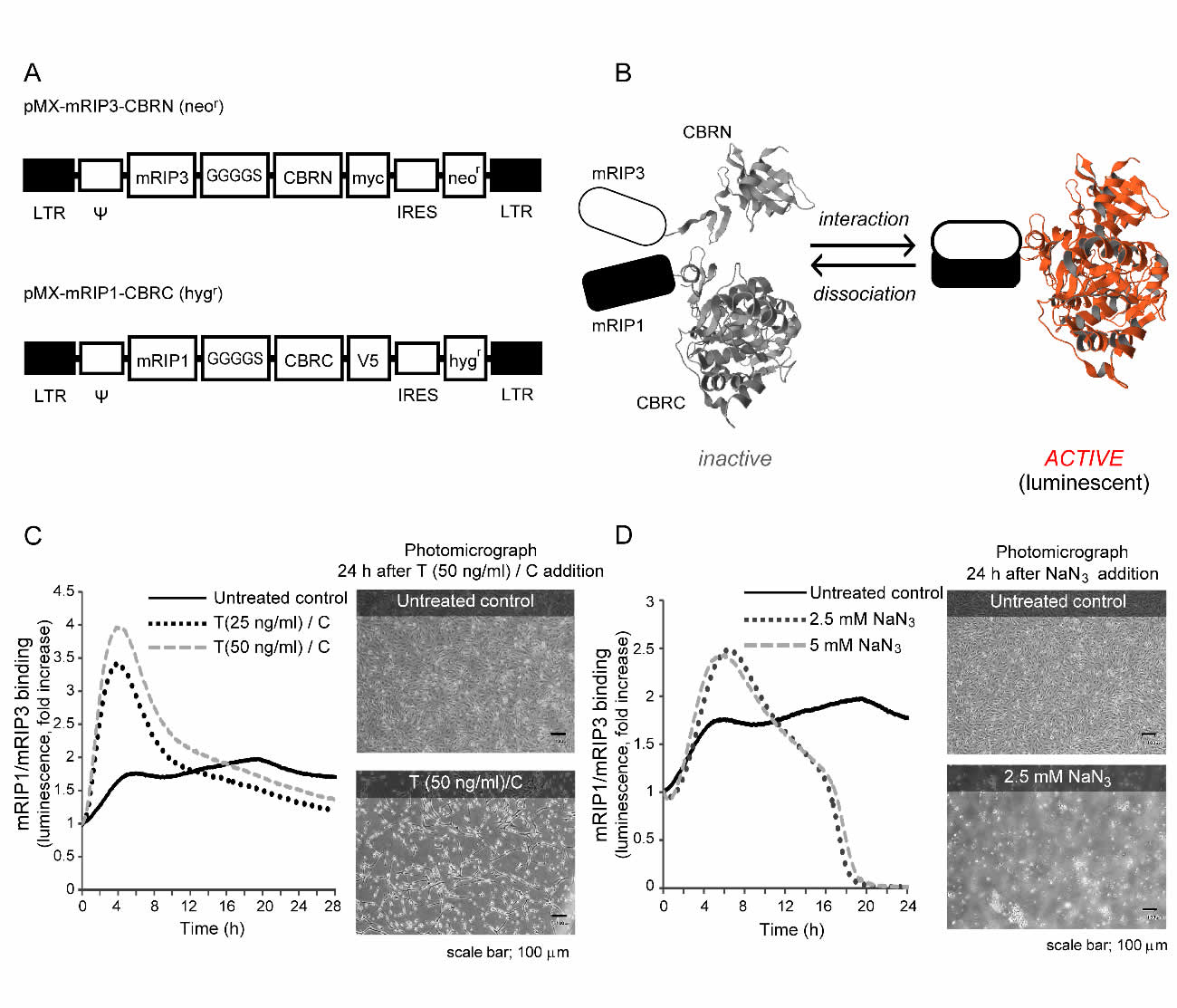
図1 cDNAベクターの構築とMEF細胞におけるTNF-α/ cycloheximideおよびSodium Azide(NaN3)による細胞死とRIP1/3結合のモニタリング
(A)cDNAベクターの構築。(B)TNF-α/ cycloheximide(T/C)は、MEF(mouse
embryonic fibroblasts)において、用量依存的にRIP1/3結合および細胞死を誘導した。(C)Sodium Azide(NaN3)投与により一時的にRIP1/3 結合は誘導されたが、その後急速に減少した。顕微鏡写真は、NaN3で処理されたMEFが大量の細胞死を起こしたことを示している。(Ref.38より引用)
TIB73マウス肝細胞株において、TNF-α/cycloheximide(T/C)はRIP1/3結合を誘導した
次に、この光プローブを導入したTIB73マウス肝細胞に対してT/Cを投与した。T/C単独投与のみではRIP1/3結合を誘導しなかったが、zVADの前投与によりRIP1/3結合は顕著に増加した(図2A)。この増加は、RIP1のアロステリック阻害剤であるNec-1により用量依存的に有意に抑制され(図2B)、かつ結合反応も遅延した。同時に、Nec-1はT/C/zVAD誘導細胞死を抑制した(図2A右の写真)。In
situ Proximity Ligation Assayにより、TIB73肝細胞におけるT/C/zVADによるRIP1/3結合とNec1による抑制を免疫細胞化学的にも確認した(図2C)。このようなT/C/zVADにより誘導されるシグナルは、Nec-1処理によって明確かつ有意に抑制された。
T/C/zVAD誘導細胞死は、RIP1/3結合を伴っていたため、T/C、zVADおよびNec-1投与後のTIB73細胞生存を経時的に調べた(図2D上パネル)。細胞生存率はT/C投与後に急速に低下したが、これはzVAD投与により部分的に改善された。このzVADによる保護効果は、投与後早期で顕著であったが、時間の経過に伴いその効果は失われた。これは、T/Cが主に投与後早期にアポトーシスを誘導していたことを示唆している。zVADによる細胞保護効果が低下した後期のタイミングでは、Nec-1が顕著かつ効果的に細胞生存を改善した。T/Cが引き起こす細胞生存率の低下に対するzVADとNec-1の独立した影響を比較すると、Nec-1は特に後期でzVADよりも細胞生存率を改善した(図2D下パネル)。これらの結果は、肝細胞においてT/Cは投与後早期に主としてアポトーシスを誘導するが、caspaseが不活性化された(反応を終えた)後期ではネクロトーシスを引き起こす可能性を示している。
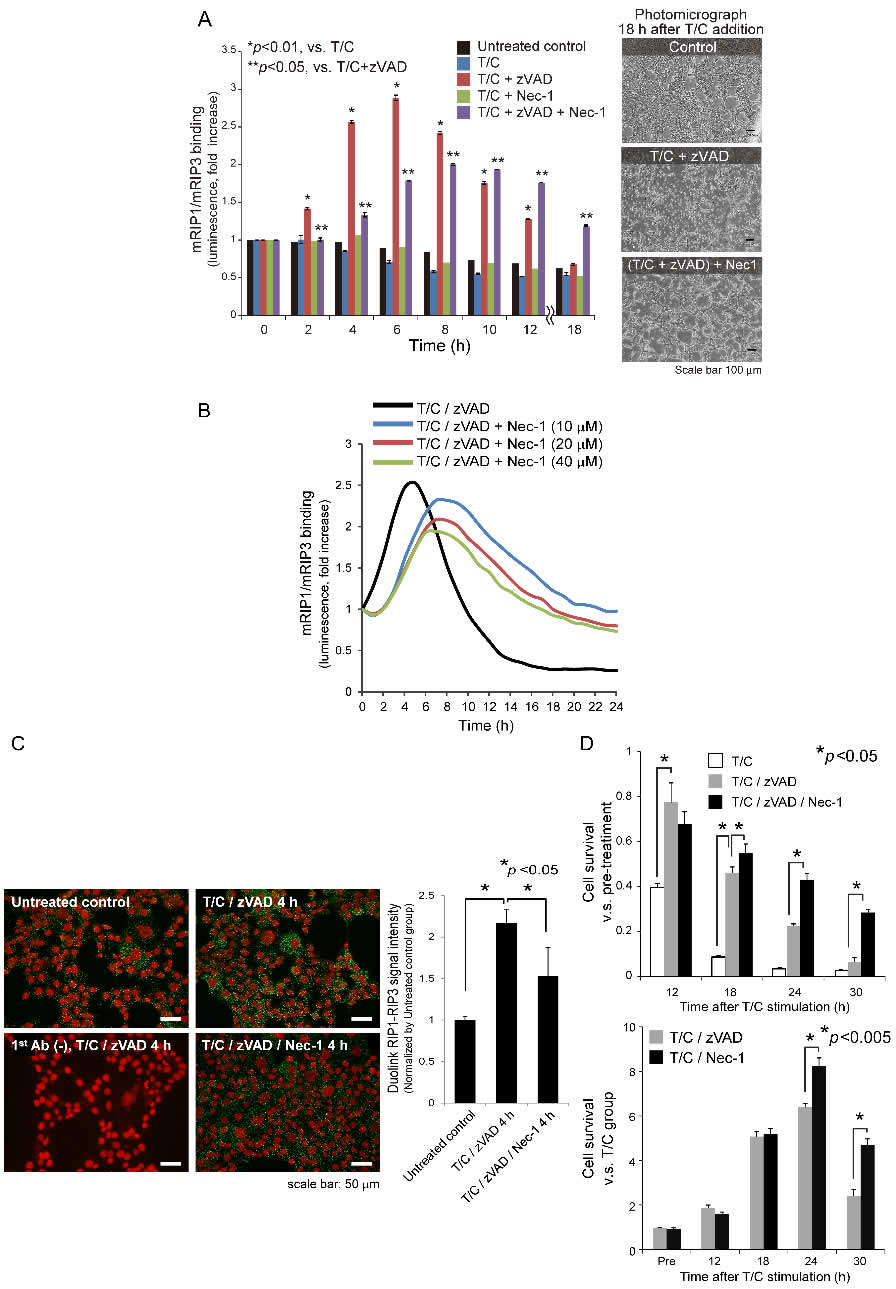
図2 TIB73マウス肝細胞において、TNF-α/ cycloheximide(T/C)はRIP1/3結合および細胞死を誘導した
(A)TIB73マウス肝細胞において、Caspase活性がz-VAD-fmk(zVAD)によって阻害された場合にのみ、T/C刺激はRIP1/3結合を誘導した。このRIP1/3結合の増加は、Necrostatin-1(Nec-1、RIP1のアロステリック阻害剤)によって抑制された。顕微鏡写真は、T/C/zVADによって誘導されたTIB73細胞における細胞死がNec-1投与によって改善されたことを示す。(B)T/C/zVADにより誘導されたRIP1/3結合は、Nec-1によって用量依存的に阻害された。(C)RIP1/3結合およびNec-1によるRIP1/3結合阻害については、免疫細胞化学的にも確認した(in situ Proximity
Ligation Assay)。赤色と緑色は、それぞれ核(DAPI; 4,6-Diamidino-2-phenylindole,
pseudo-color)とRIP1/3結合を示す。(D)T/C処理後に細胞生存率が急激に低下し、これはzVAD投与により部分的に改善された。T/C/zVAD細胞死(上図)とT/C細胞死(下図)は共にNec-1を加えることで有意に改善された(特にcaspaseが不活性化した場合)。これは、T/C刺激が後のタイミングでネクロトーシスを誘発することを示唆した。(Ref.38より引用)
TIB73マウス肝細胞において、Fas刺激はアポトーシスとネクロトーシス両者を誘導した
次に、マウス肝細胞におけるFasL/Fasによるプログラム細胞死の誘導について検討した。FasL刺激は著明な細胞死を誘導したが、zVAD投与ではその細胞死を部分的に阻害したのみであった(図3A)。これは、FasLが誘導する肝細胞死において、アポトーシスとは異なる細胞死が関与していることを示唆している。そこで、それぞれアポトーシスとネクロトーシスを表す“caspase3活性の光プローブと“RIP1/3結合光プローブを用いて、FasLが誘導する細胞死におけるこれら2種類の細胞死を動的に解析した(図3B)。FasLは投与後4~6時間でcaspase3の速やかな活性化を引き起こしたが、RIP1/3結合は誘導しなかった。しかし、zVADを同時投与することでRIP1/3の結合は速やかに増加した(図3B右パネル)。これらの結果は、ネクロトーシスがアポトーシスの「バックアップ」として、caspase依存経路が利用できない場合に(または活性化後)、細胞死を達成するという従来の仮説を支持するものとなった17,23。
肝細胞において、TNF-αとFasLは共にレドックス依存的に細胞死を誘導することが知られている5,7,35,36。肝細胞におけるリガンド誘導性ネクロトーシスへの酸化ストレスの関与を確認するために、FasL誘導性ネクロトーシスに対する抗酸化剤の効果を調べた。抗酸化剤(TroloxおよびNAC)を前投与しておくと、FasLにより誘導されるRIP1/3結合は有意に抑制された(図3C)。このことは、酸化ストレスが肝細胞におけるネクロトーシス誘導のプロセスに関与していることを示している。
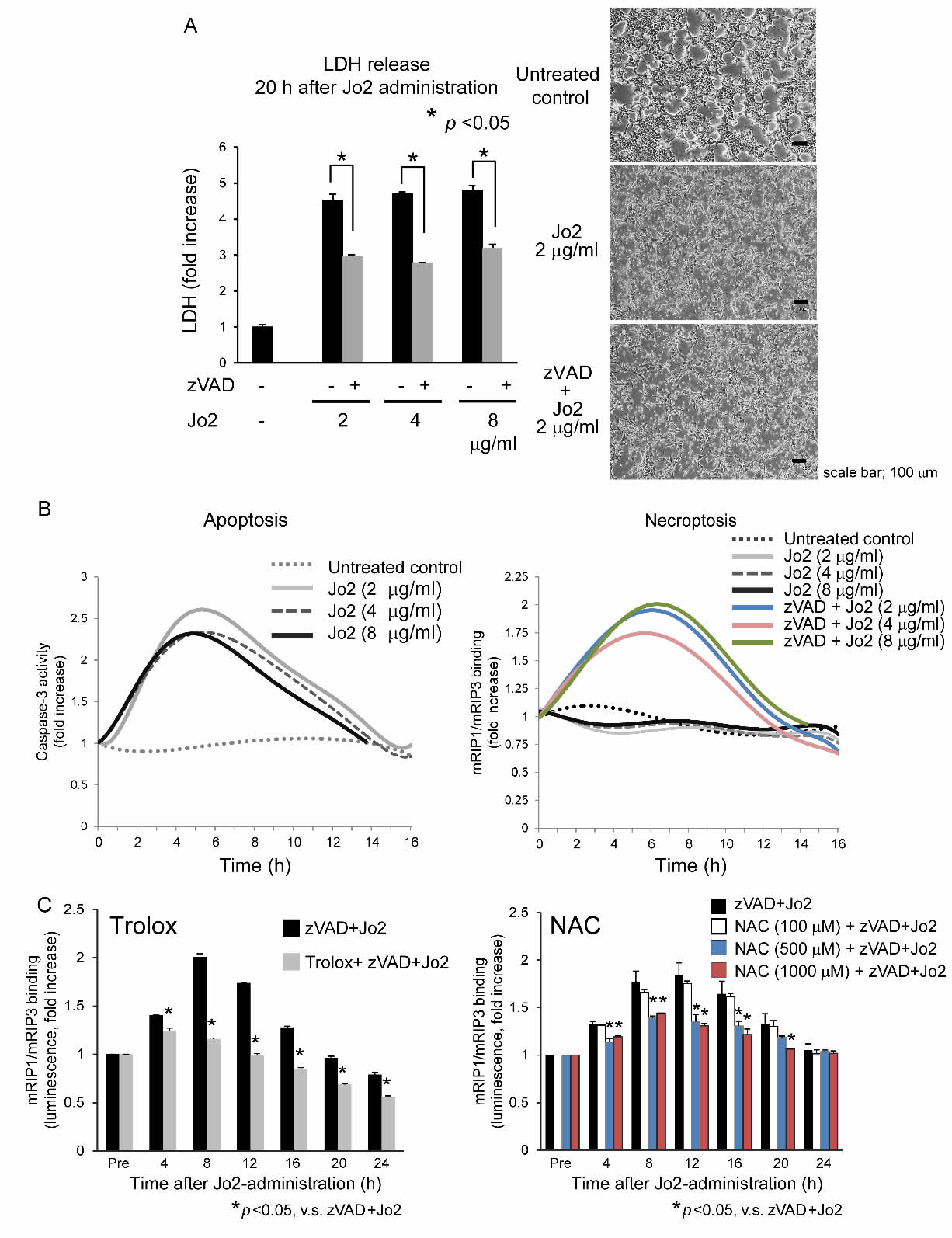
図3 TIB73肝細胞において、FasL(Jo2)はレドックス依存的に細胞死およびRIP1/3結合を誘導した
(A)caspase阻害剤(zVAD)は、FasL(Jo2)が誘導する細胞死を部分的に抑制した。(B)光プローブを用いて、caspase-3活性(アポトーシス、左図)およびRIP1/3結合(ネクロトーシス、右図)を評価した。FasL(Jo2)はcaspase-3を活性化し、同時にcaspaseが不活性化された場合にのみRIP1/3結合を誘導した。(C)抗酸化剤(TroloxおよびN-acetyl cysteine [NAC])は、FasL(Jo2)が誘導したRIP1/3結合を有意に抑制した。(Ref.38より引用)
TIB73マウス肝細胞において、低酸素・再酸素化刺激はレドックス依存的にネクロトーシスを誘導する
次に、マウス肝細胞において、低酸素・再酸素化(H/R)がネクロトーシスを誘導するかどうかを検討した。H/Rは、再酸素化後16〜18時間でRIP1/3結合を増加させ、細胞死を誘導したが(図4A)、それらはいずれもNec-1によって有意に抑制された。
また、H/Rにより誘導されたRIP1/3結合は、抗酸化剤(TroloxおよびNAC)によって用量依存的かつ有意に阻害された(図4B)。これらのデータは、H/RがFasLと同様に、レドックス依存的にRIP1/3結合(すなわちネクロトーシス)を誘導することを示している。
活性酸素(H2O2)それ自体でも肝細胞内のRIP1/3結合を誘導した。興味深いことに、H2O2により誘導されたRIP1/3結合はNec-1によって阻害されたが、zVADの影響は受けなかった(図4C)。
内因性のネクロトーシス関連タンパク(RIP1、RIP3およびMLKL)の発現レベルは、低酸素後一時低下したが、RIP1とMLKLの発現は、16時間後から回復し始めた。RIP3はH/R後16時間後に一時的に回復したが、その後、再度減少した(図4D)。
H/RによるRIP1/3結合の誘導は、FasLで刺激した場合より遅延したが、FasLとは異なりcaspase阻害によって増加せず、むしろ若干減少した(図3Bおよび4E)。このことは、H/R-およびFasL/Fasによるネクロトーシス誘導において、相異なる分子メカニズムの存在可能性を示唆している。肝細胞において、FasLおよびH/Rによるネクロトーシスの誘導は、各々caspase依存的にあるいはcaspase非依存的に生じたが、どちらも共にレドックス依存的であった。

図4 TIB73肝細胞において、低酸素/再酸素化刺激はレドックス依存的に RIP1/3 結合および細胞死を誘導した
(A)低酸素/再酸素化(H/R)はRIP1/3結合にそれに伴う細胞死を誘導したが、Nec-1により阻害された。(B)H/Rが誘導するRIP1/3結合は、抗酸化剤(TroloxおよびNAC)により用量依存的に阻害された。(C)H2O2により誘導されたRIP1/3結合はNec-1投与により有意に阻害されたが、FasL投与時とは対照的にcaspase阻害によって増強されることはなかった。(D)低酸素によりネクロトーシス関連タンパク(RIP1、RIP3、MLKL)の発現は一過性に低下したが、再酸素化後徐々に回復した。これは、(A)に示すRIP1/3結合のタイミングと一致した。(E)H/Rが誘導する RIP1/3結合はcaspase阻害によって増強されなかった。この結果はH2O2処理の結果と同様であった。(Ref.38より引用)
課題と展望
今回の研究では、光プローブの開発過程ではMEFを用いた。肝細胞における解析に関しては、今回技術的な問題でTIB73マウス胎児肝細胞株を用いた。この細胞をもちいた実験結果は、これまでの報告と矛盾するものではないが、これをもって直ちに成体肝細胞の特徴として考えることは難しい。今後AML12などの細胞をもちいた実験あるいはマウスを用いた実験により検証する必要があると考えている。
生命現象の中で、caspase活性(特にcaspase8活性)が抑制される特殊な病態生理学的な状況に関して具体的にあまり知られていない。しかしながら、caspaseの活性化は発生・分化を含めた様々な生命現象が起こる過程において重要な役割を果たしている。適切なタイミングで必要な細胞死を誘導し生物学的に必要なプロセスを遅延なく進めることは重要であり、caspaseが活性化できない場合に代替的に活性化するシグナル経路の存在は、アポトーシスの重要なバックアップ機構とも考えられている。さらに、今回の検討では、caspaseに依存せずレドックスに依存しネクロトーシスのプロセスが進行することが確認された。今後、caspase依存性あるいはcaspase非依存性に進行するネクロトーシスのより詳しい検討により、様々な病態の進行過程の中でのこの経路の生理病態学的な重要性39が示される必要がある。
光プローブの適切な利用は、細胞内・生体内での様々なserendipitousな発見と既知の現象の相互関係の理解を助けるものと期待する。今回のようにcaspase3活性とRIP1/3結合の光プローブを組み合わせて使用することは、細胞内イベントの進行過程をより正確に観察したり、あるいは相互作用を確認したりするのに有益であると思われる。どのような分子機能を光プローブで見るのが適切か、あるいは光プローブ導入により細胞内イベントに影響を与えてしまう可能性があるなど種々の課題はあるものの、従来の手法で見いだされた知見を、生細胞・生体組織でよりダイナミックに理解する一助になると考えている。一方、我々は光を利用した細胞機能操作の実験も進めているが、こういった試みは(一旦プローブ遺伝子を安定的に導入した後は)細胞・生体をできるだけ非侵襲的な方法で観察あるいは操作することで、より正確に理解することを可能とし、今後さらなる発展が期待される。
終わりに
肝細胞において、caspaseが不活性化された場合には T/Cおよび FasLはネクロトーシスを誘導する。また、FasLおよびH/Rが誘導するネクロトーシスは、レドックスに依存している。低酸素・再酸素化は、活性酸素(reactive oxygen
species, ROS)に依存するメカニズムでネクロトーシスを誘導するが、caspase活性に依存しないメカニズムの関与も考えられる(図5)。肝細胞におけるネクロトーシス誘導の正確なメカニズムは未だ不明であり、さらなる研究が必要である。
光プローブを用いたこのような解析は、分子病態メカニズムをダイナミックに理解し、また細胞・小動物レベルにおけるより詳細な検討にも有用と考えられる。
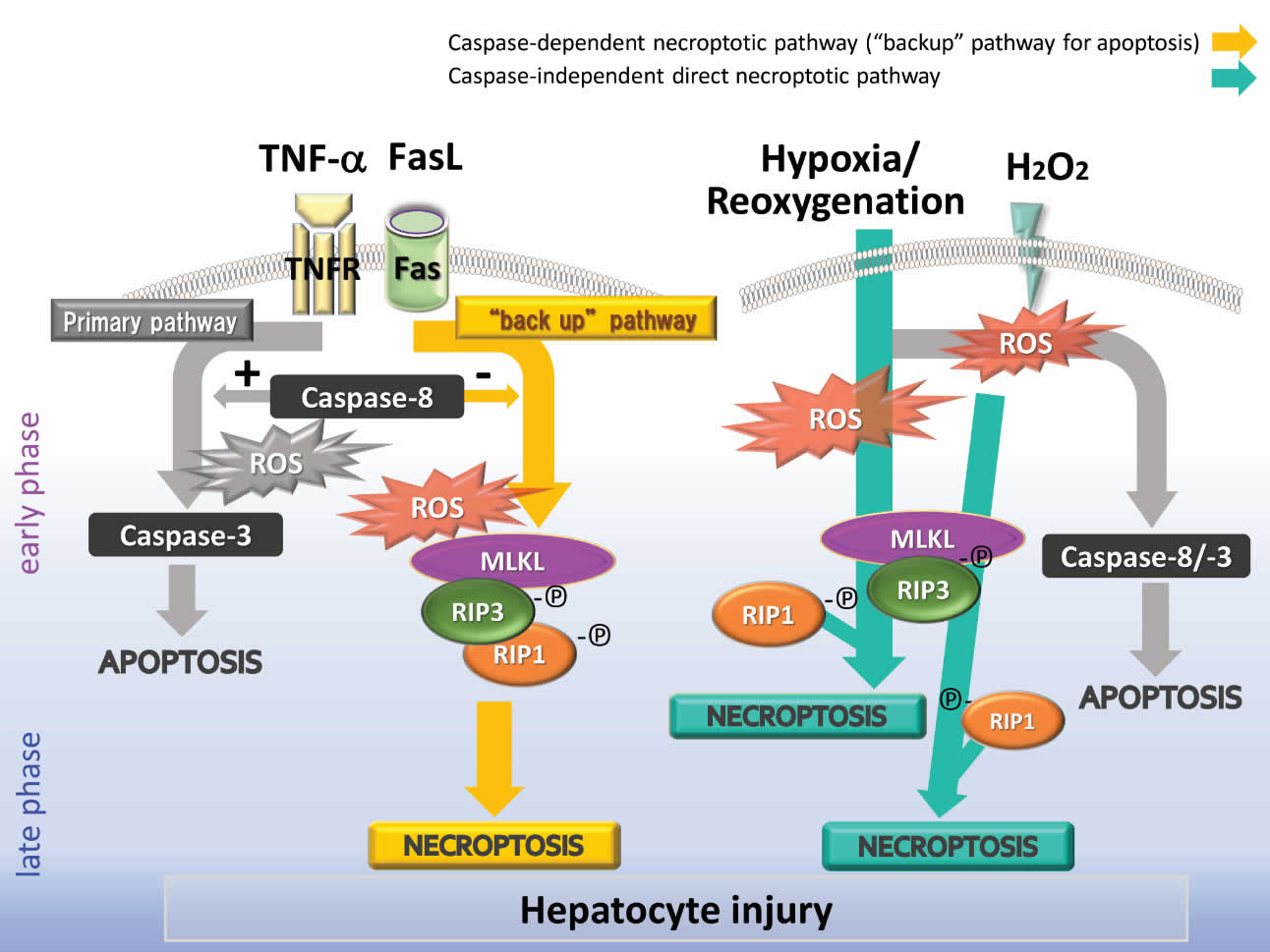
図5 レドックスが関与するプログラム細胞死(アポトーシスおよびネクロトーシス)
肝細胞において、TNF-α/FasLは主にcaspase8/-3経路を介してレドックスに依存してアポトーシスを誘導する。Caspase-8が利用できない(あるいは、不活性化されている)と、代替経路が活性化されネクロソーム(MLKL/RIP1/RIP3)を形成し、レドックス依存的にネクロトーシスを誘導する。ネクロトーシスは、アポトーシスよりも遅いタイミングで発生し、アポトーシスの“バックアップ機構”としての役割を果たしている可能性がある。活性酸素reactive oxygen
species; ROS, 例えばH2O2)は、主にMLKLの存在下でアポトーシスとネクロトーシスを誘導する。H/Rによるネクロトーシスの誘導は、MLKL、RIP1およびRIP3タンパクの再酸素化後の再発現により強く影響されている可能性があるが、caspase活性はこれに関与しない。(Ref.38より引用)
REFERENCES
- Dunn W, Shah VH. Pathogenesis of Alcoholic Liver Disease. Clin Liver Dis. 2016;20:445-56.
- Khoury T, Rmeileh AA, Yosha L, Benson AA, Daher S, Mizrahi M. Drug Induced Liver Injury: Review with a Focus on Genetic Factors, Tissue Diagnosis, and Treatment Options. J Clin Transl Hepatol. 2015;3:99-108.
- Oh IS, Park SH. Immune-mediated Liver Injury in Hepatitis B Virus Infection. Immune Netw. 2015;15:191-8.
- Suhail M, Abdel-Hafiz H, Ali A, Fatima K, Damanhouri GA, Azhar E, Chaudhary AG, Qadri I. Potential mechanisms of hepatitis B virus induced liver injury. World J Gastroenterol. 2014;20:12462-72.
- Haga S, Ozawa T, Yamada Y, Morita N, Nagashima I, Inoue H, Inaba Y, Noda N, Abe R, Umezawa K, Ozaki M. p62/SQSTM1 plays a protective role in oxidative injury of steatotic liver in a mouse hepatectomy model. Antioxid Redox Signal 2014;21:2515-30.
- Haga S, Remington SJ, Morita N, Terui K, Ozaki M. Hepatic Ischemia Induced Immediate Oxidative Stress after Reperfusion and Determined the Severity of the Reperfusion-induced Damage. Antioxid Redox Signal. 2009;11:2563-72.
- Haga S, Terui K, Zhang HQ, Enosawa S, Ogawa W, Inoue H, Okuyama T, Takeda K, Akira S, Ogino T, Irani K, Ozaki M. Stat3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J Clin Invest. 2003;112:989-98.
- Bahirwani R, Reddy KR. Drug-induced liver injury due to cancer chemotherapeutic agents. Semin Liver Dis. 2014;34:162-71.
- Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J Gastroenterol Hepatol. 2011;26:173-9.
- Wang K. Molecular mechanisms of liver injury: apoptosis or necrosis. Exp Toxicol Pathol. 2014;66:351-6.
- Gao WY, Li D, Cai DE, Huang XY, Zheng BY, Huang YH, Chen ZX, Wang XZ. Hepatitis B virus X protein sensitizes HL-7702 cells to oxidative stress-induced apoptosis through modulation of the mitochondrial permeability transition pore. Oncol Rep. 2017;37:48-56.
- Pinkoski MJ, Brunner T, Green DR, Lin T. Fas and Fas ligand in gut and liver. Am J Physiol Gastrointest Liver Physiol. 2000;278:G354-6.
- Galle PR, Hofmann WJ, Walczak H, Schaller H, Otto G, Stremmel W, Krammer PH, Runkel L. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med. 1995;182:1223–30.
- Kanzler S, Galle PR. 2000. Apoptosis and the liver. Semin Cancer Biol. 2000;10:173–84.
- Faubion WA, Guicciardi ME, Miyoshi H, Bronk SF, Roberts PJ, Svingen PA, Kaufmann SH, Gores GJ. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest. 1999;103:137–45.
- Castaneda F, Kinne RK. Ethanol treatment of hepatocellular carcinoma: high potentials of low concentrations. Cancer Biol Ther. 2004;3:430-3.
- Saeed WK, Jun DW. Necroptosis: an emerging type of cell death in liver diseases. World J Gastroenterol. 2014;20:12526-32.
- Shen B, He Y, Zhou S, Zhao H, Mei M, Wu X. TRPC6 May Protect Renal Ischemia-Reperfusion Injury Through Inhibiting Necroptosis of Renal Tubular Epithelial Cells. Med Sci Monit. 2016;22:633-41.
- Linkermann A, De Zen F, Weinberg J, Kunzendorf U, Krautwald S. Programmed necrosis in acute kidney injury. Nephrol Dial Transplant. 2012;27:3412-9.
- Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, Zhang M, Wu H, Guo J, Zhang X, Hu X, Cao CM, Xiao RP. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175-82.
- Sendler M, Mayerle J, Lerch MM. Necrosis, Apoptosis, Necroptosis, Pyroptosis: It Matters How Acinar Cells Die During Pancreatitis. Cell Mol Gastroenterol Hepatol. 2019;2:407-8.
- Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 147 (2014) 765-783.
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455-65.
- Naomoto Y, Tanaka N, Fuchimoto S, Orita K. In vitro synergistic effects of natural human tumor necrosis factor and natural human interferon-alpha. Jpn J Cancer Res. 1987;78:87-92.
- Fulda S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol Ther. 2013;14:999-1004.
- Onishi M, Kinoshita S, Morikawa Y, Shibuya A, Phillips J, Lanier LL, Gorman DM, Nolan GP, Miyajima A, Kitamura T. Applications of retrovirus-mediated expression cloning. Exp Hematol. 1996;24:324-9.
- Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063-6.
- Crawford LE, Milliken EE, Irani K, Zweier JL, Becker LC, Johnson TM, Eissa NT, Crystal RG, Finkel T, Goldschmidt-Clermont PJ. Superoxide-mediated actin response in post-hypoxic endothelial cells. J Biol Chem. 1996;271:26863–7.
- Ozaki M, Deshpande SS, Angkeow P, Suzuki S, Irani K. Rac1 regulates stress-induced, redox-dependent heat shock factor activation. J Biol Chem. 2000, 275:35377-83.
- Ozaki M, Haga S, Ozawa T. In vivo monitoring of liver damage by caspase-3 probe. Theranostics 2012;2:207-14.
- Sorokina EM, Feinstein SI, Zhou S, Fisher AB. Intracellular targeting of peroxiredoxin 6 to lysosomal organelles requires MAPK activity and binding to 14-3-3ε. Am J Physiol Cell Physiol. 2011;300:C1430-41.
- Petri MK, Koch P, Stenzinger A, Kuchelmeister K, Nestler U, Paradowska A, Steger K, Brobeil A, Viard M, Wimmer M. PTPIP51, a positive modulator of the MAPK/Erk pathway, is upregulated in glioblastoma and interacts with 14-3-3β and PTP1B in situ. Histol Histopathol. 2011;26:1531-43.
- Jin S, Ray RM, Johnson LR. TNF-αlpha/cycloheximide-induced apoptosis in intestinal epithelial cells requires Rac1-regulated reactive oxygen species. Am J Physiol Gastrointest Liver Physiol. 2008;294:G928-37.
- Ji D, Kamalden TA, del Olmo-Aguado S, Osborne NN. Light- and sodium azide-induced death of RGC-5 cells in culture occurs via different mechanisms. Apoptosis. 2011;16:425-37.
- Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-αlpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583-9.
- Kastl L, Sauer SW, Ruppert T, Beissbarth T, Becker MS, Süss D, Krammer PH, Gülow K. TNF-α mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-κB activation in liver cells. FEBS Lett. 2014;588:175-83.
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, Yang Z, Wu SQ, Chen L, Han J. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994-1006.
- Sanae Haga, Akira Kanno, Takeaki Ozawa, Naoki Morita, Mami Asano, Michitaka Ozaki. Detection of Necroptosis in Ligand-Mediated and Hypoxia-Induced Injury of Hepatocytes Using a Novel Optic Probe-Detecting Receptor-Interacting Protein (RIP) 1/RIP3 Binding. Oncol. Res. 2018; 26 (3) : 503-513.
- Sanae Haga, PhD, Akira Kanno, PhD, Naoki Morita, PhD, Shigeki Jin, PhD, Kotaro Matoba, MD, Takeaki Ozawa, PhD, Michitaka Ozaki, MD, PhD. Poly (ADP-ribose) polymerase (PARP) is critically involved in liver ischemia/reperfusion-injury. J Surg Res 2022; 270: 124-138.

