





研究交流
肝傷害後の組織修復におけるストレス関連キナーゼMKK7の役割
大塩貴子1,2、西川祐司1
1 旭川医科大学病理学講座腫瘍病理分野
2 北海道大学遺伝子病制御研究所がん制御学分野(現所属)
はじめに
JNK-c-Jun経路は細胞外からのストレスや炎症性サイトカインに応答し、細胞の増殖・生存・分化などを制御する (1)。JNKの活性化には上流キナーゼであるMitogen-activated protein kinase kinase(MKK)7とMKK4が必須であり、これらのいずれかをマウスでノックアウト(KO)すると、肝形成が強く阻害され、胎生致死となる (2-5)。一方、成熟マウス肝でMKK4をノックダウンすると、MKK7の活性化を介して肝細胞増殖や肝再生が促進されることが報告され、肝細胞におけるMKK7の役割が注目されていた (6)。そこで、我々は成熟肝細胞におけるMKK7の役割を解明するために、Mkk7-floxマウスをAlb-CreまたはMx1-Creマウスと掛け合わせて肝細胞のMKK7をKOし、解析を行った。その結果、MKK7 KOは初代培養肝細胞の増殖や部分肝切除後の肝再生に影響を与えなかったが、四塩化炭素(CCl4)投与による急性傷害からの肝修復を遅延させることが明らかになった (7)。そこで本稿では、傷害後の肝修復機構におけるMKK7の役割について報告する。
MKK7 KOは肝傷害後の組織修復を遅延させる
まず我々はMKK7 KOによる肝傷害後の組織修復への影響を調べるために、MKK7 KO(Mx1-Cre)および対照マウスに四塩化炭素(1 mL/kg)を皮下投与した。四塩化炭素は中心静脈肝周辺の肝細胞によって代謝されて小葉中心性の肝細胞壊死を引き起こすが、肝傷害の指標である血漿中のalanine aminotransferase(ALT)や肝細胞増殖、マクロファージの集積、各種炎症性サイトカインの発現は対照群とMKK7 KO群において同程度であった(図1A, B)(7)。しかし、MKK7 KO肝では、傷害4日後に小葉中心部の壊死組織がより多く残存し、対照肝で修復がほぼ完了する8日後においても傷害部が観察された(図1C)。四塩化炭素による肝細胞壊死部にはfibrinが蓄積し、傷害部はfibrinogen抗体を用いた免疫組織化学によって染色される (8)。Fibrinogen陽性部の面積は、傷害2日後の肝臓においてはMKK7 KOと対照に差はなかったが、傷害4日後ではMKK7 KO群において有意に増加していた。
以上の結果より、MKK7は成熟肝細胞の増殖には関わらないが、傷害後の肝組織修復に重要な役割を果たしていることが示唆された。また、この現象は、Mx1-Creシステムによる肝細胞と血液細胞特異的KOとAlb-Creシステムによる肝細胞と小葉間胆管細胞特異的なKOの両方において観察されており (7)、肝細胞のMKK7が組織修復に関与していると考えられた。
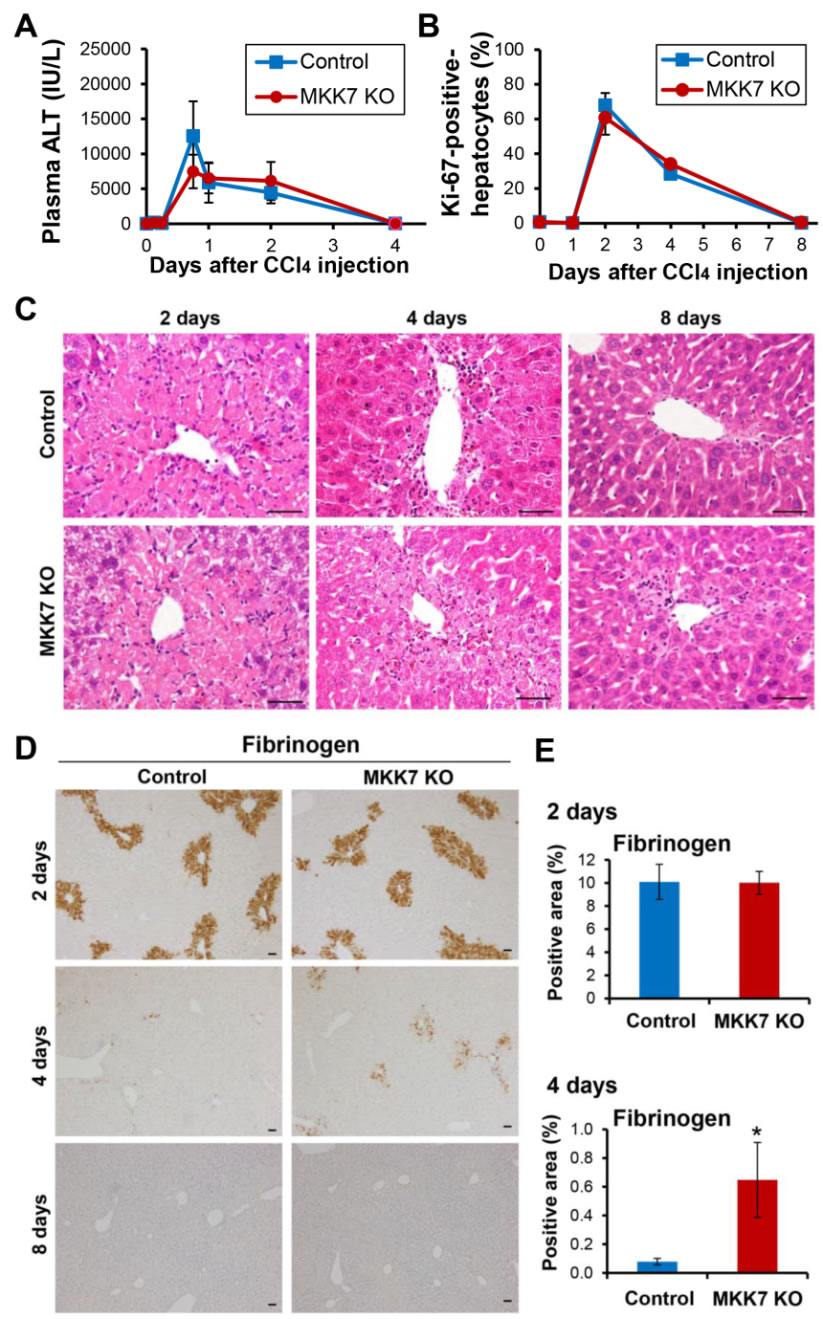
図1.MKK7 KO(Mx1-Cre)マウスにおける四塩化炭素(CCl4)傷害後の肝組織修復の遅延
(A) 四塩化炭素投与後の血漿中のalanine aminotransferase(ALT)の変化。
(B) 四塩化炭素投与後のKi-67陽性肝細胞の割合。
(C) 四塩化炭素投与後の肝傷害部の組織像(HE染色)。Scale bar, 50 μm.
(D) 四塩化炭素投与後の肝組織でのfibrinogen免疫組織化学。Scale bar, 50 μm.
(E) 四塩化炭素投与後のfibrinogen陽性領域の割合。*P < 0.05 versus control.(文献7より改編引用)
MKK7はTagln、Glipr2、Plauの発現を介してコラーゲンゲル内での樹枝状形態形成を促進する
肝細胞でのMKK7 KOの影響を明らかにするために、対照およびMKK7 KOマウス(Alb-Cre)より肝細胞を分離し、肝細胞スフェロイドをI型コラーゲン内に包埋する三次元培養を行った (9)。肝細胞スフェロイドはEGFとインスリンの存在下で樹枝状の細胞突起を伸ばし、この反応はTNF-αやTGF-βを添加することによりさらに促進された(図2)。一方、MKK7 KO肝細胞スフェロイドは突起の伸長が弱く、TNF-αやTGF-βに対する反応も抑制されていた(図2)。
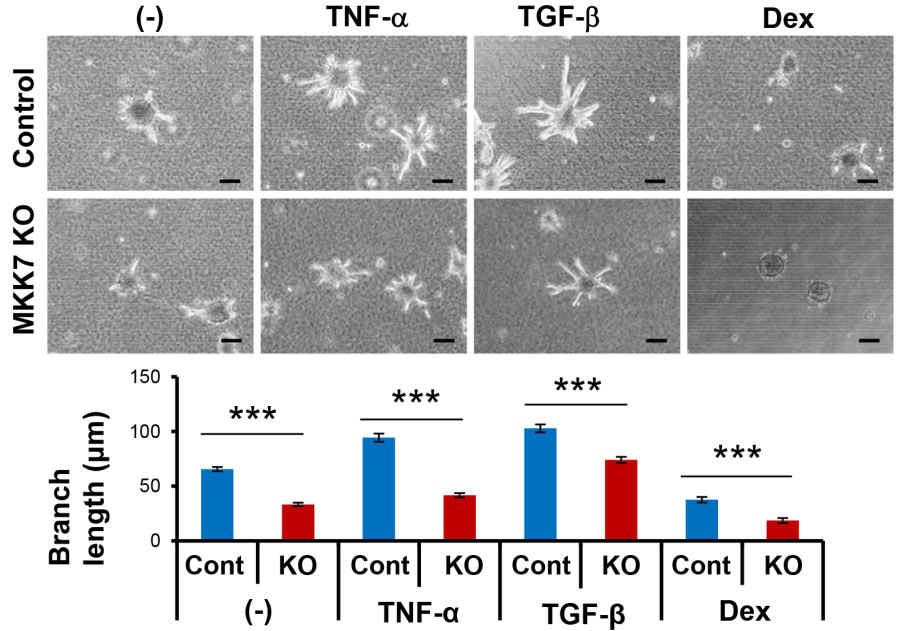
図2.MKK7 KO(Alb-Cre)による肝細胞スフェロイドの樹枝状突起形成の抑制
肝細胞スフェロイドのI型コラーゲンゲル内での培養2日後の位相差像と樹枝状突起の定量。Scale bar, 100 μm. 樹枝状突起形成はTNF-αやTGF-βの添加によって促進され、dexamethasone(Dex)によって抑制されたが、MKK7 KOは全ての条件において対照よりも突起の伸長を抑制した。***P < 0.001(文献7より改編引用)
そこで我々はこの反応に関連する遺伝子を特定するために、マイクロアレイ解析を行い、その遺伝子発現パターンの中から、樹枝状突起の伸長パターンと同様な発現をし、これまでに細胞運動に関与するとの報告のある遺伝子について絞り込んだ(図3A)(10-12)。その結果、コラーゲンゲル培養での形態形成能に関与する候補遺伝子としてtransgelin(Tagln)、glioma pathogenesis related-2(Glipr2)、plasminogen activator urokinase-type(uPA)(Plau)が同定された(図3A)。さらに、MKK7 KO肝細胞にアデノ随伴ウイルス(AAV8)を用いてこれらの遺伝子を過剰発現させると、コラーゲン内での形態形成能が回復した(図3B)。
以上より、MKK7 KO肝細胞ではTagln、Glipr2、Plauの発現低下により細胞外マトリクスを介した細胞運動が抑制されることが示唆された。さらに、野生型の肝細胞スフェロイドをJNK阻害剤で処置するとコラーゲンゲル内での樹枝状形態形成が抑制され、TaglnとPlauの発現が低下したが、Glipr2の発現に影響はなかった (7)。そこで、我々はこれら3つの候補遺伝子のうち、JNKシグナルの下流で発現誘導されるTaglnとPlauに焦点を当て、検討を行った。
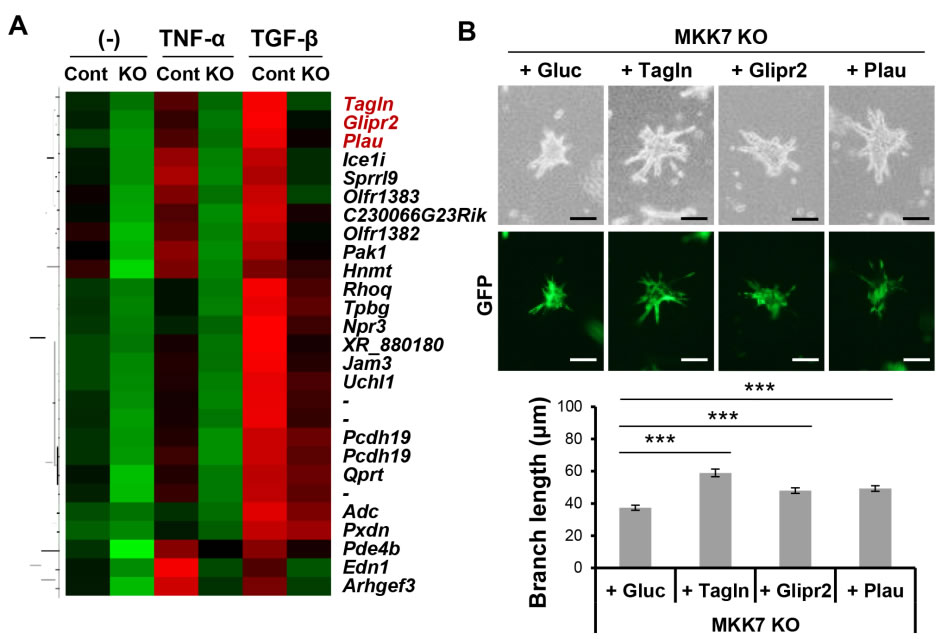
図3.Tagln、Glipr2、Plauの発現低下によるMKK7 KO(Alb-Cre)肝細胞での樹枝状突起形成の抑制
(A) コラーゲンゲル培養2日後の肝細胞において、樹枝状突起の伸長と似たパターンを示す遺伝子発現のクラスター解析。これらの樹枝状形態形成に関与する遺伝子候補のうち、細胞運動に関与することが報告されているTagln、Glipr2、Plauに着目した。
(B) Gluc(対照)、Tagln、Glipr2、Plau過剰発現MKK7 KO肝細胞のスフェロイド培養2日目の位相差像、GFPの蛍光、樹枝状突起の定量。GFP陽性細胞は上記の遺伝子を共発現するアデノ随伴ウイルス(AAV8)ベクターの導入された細胞を示す。Gluc発現細胞に比べ、Tagln、Glipr2、Plau発現細胞では有意に樹枝状突起の伸長が亢進した。***P < 0.001. Scale bar, 50 μm.(文献7より改編引用)
Plauの強制発現はMKK7 KOによる肝修復遅延を解消する
上記で同定した候補遺伝子がMKK7 KOによる肝傷害後の修復遅延に関与しているかを調べるために、肝臓特異的MKK7 KOマウスの肝臓にAAV8を用いてTaglnとPlau、uPAに関連するplasminogen activator tissue-type(PA)(Plat)を過剰発現させ、四塩化炭素投与による肝傷害を誘導した。肝臓にPlauを発現させると肝傷害を引き起こすことが報告されているように (13, 14)、Plauを過剰発現させた肝組織では軽度の傷害が観察されたが、四塩化炭素投与前および1日後の血中ALTレベルは、対照やそれぞれの遺伝子を過剰発現させたものと有意な差は見られなかった(図4C)。さらに、四塩化炭素投与2日後でのfibrinogen陽性領域の面積はそれぞれの群で同様であったが、4日後にはPlauを過剰発現させたマウスにおいて顕著に面積が減少していた(図4A, B, D)。これらの結果より、MKK7 KOによる四塩化炭素傷害の回復の遅延は、少なくとも一部はuPAの発現低下に起因することが示唆された。
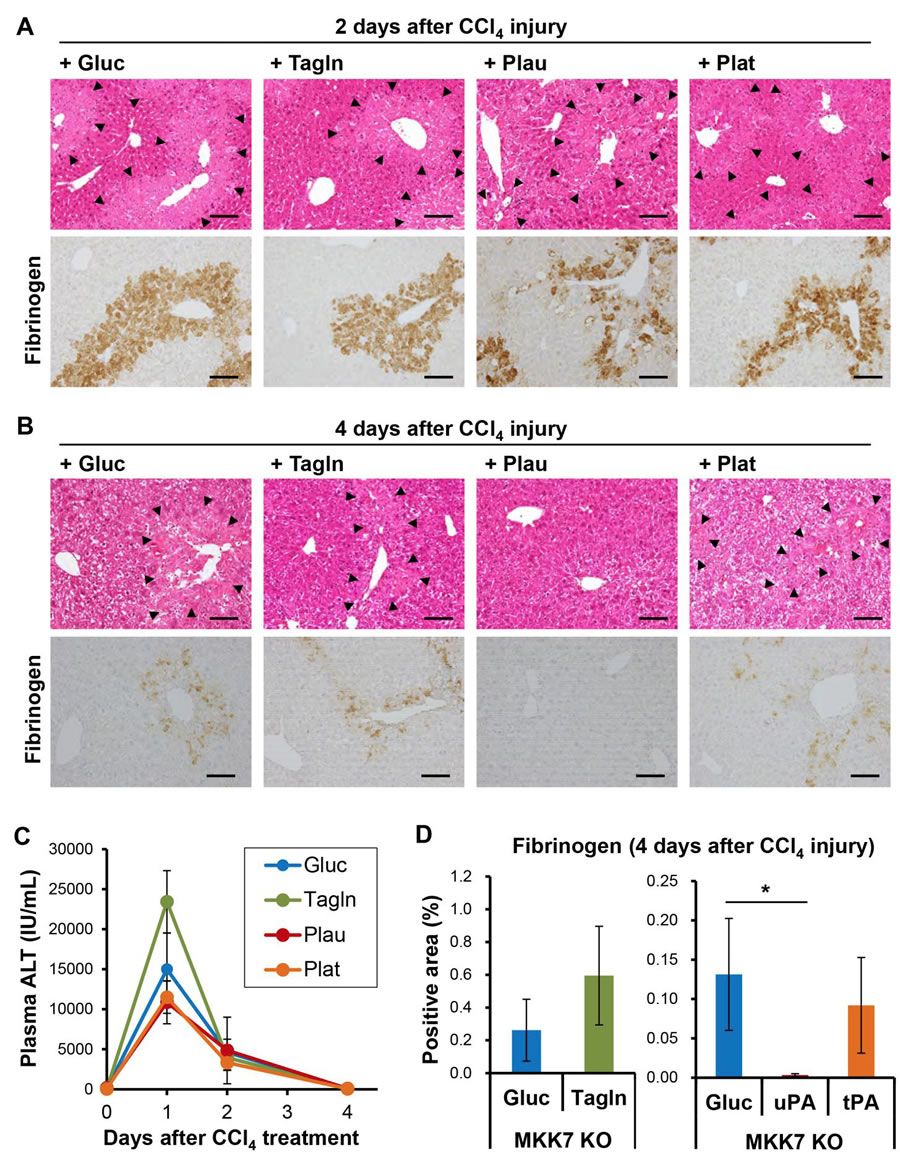
図4. Plau過剰発現による四塩化炭素傷害後のMKK7 KO(Alb-Cre)肝組織修復遅延の回復
(A, B) 肝臓特異的MKK7 KOマウスにAAV8を用いてGluc(対照)、Tagln、Plau、Platを過剰発現させ、四塩化炭素投与後2日 (A) および4日 (B) の肝組織像(HE染色とfibrinogen免疫組織化学)。矢頭で囲まれた部分は傷害領域を示す。Scale bar, 50 μm.
(C) 四塩化炭素投与後の血漿中のALTの変化。
(D) 四塩化炭素投与後4日の肝組織におけるfibrinogen陽性領域の割合。*P < 0.05.(文献7より改編引用)
おわりに
肝細胞におけるMKK4のノックダウン実験により、MKK7が成熟マウスの肝細胞増殖に重要であることが示唆されていたが (6)、我々は、MKK7 KOが肝細胞の増殖に影響しないことを見出した。一方、MKK7はPlauの発現を介して傷害を伴う肝再生が適切に行われるために重要な役割を担っていることが示唆された(図5)。実際に、Plauやそれによって活性化されるplasminogenのKOマウスでは、四塩化炭素傷害後の組織修復が著しく障害される (15, 16)。
全身性のMKK7 KOマウスは、E9.5までに異常はみられないが、E11.5において肝形成が顕著に阻害される (2-5)。しかし、本研究により、Alb-CreによるMKK7 KOは肝発生に影響しないことが明らかになった。Albの発現はE10.5に肝臓原基で検出され (17)、Alb-Creによる遺伝子組み換えはその後数日で完了する (18)。したがって、E10.5以前の肝芽細胞もしくは胎生期での非実質細胞でのMKK7発現が肝発生に重要である可能性が考えられた。

図5. MKK7の肝組織修復における役割(模式図)
組織破壊を伴う肝傷害により、肝細胞においてMKK7が強く誘導され、Plau、Tagln、Glipr2などの遺伝子発現が亢進する。これらの遺伝子産物は肝細胞の移動や運動に影響を与えるだけでなく、特にuPAの発現を介して細胞外マトリクスを分解し、組織修復に寄与していることが推察される。
謝辞
本稿で紹介させていただいた研究は、旭川医科大学病理学講座腫瘍病理分野と東京医科歯科大学難治疾患研究所発生再生生物学分野の仁科博史先生、神戸大学医学研究科分子細胞生物学分野の鈴木聡先生、ブリティッシュコロンビア大学生命科学研究所遺伝医学講座のJosef Penninger先生との共同研究です。本研究の途中経過は肝細胞研究会でも発表させていただいており、ご助言いただいた先生方に感謝申し上げます。
参考文献
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000;103:239-252.
- Ganiatsas S, Kwee L, Fujiwara Y, Perkins A, Ikeda T, Labow MA, Zon LI. SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc Natl Acad Sci U S A 1998;95:6881-6886.
- Nishina H, Vaz C, Billia P, Nghiem M, Sasaki T, De la Pompa JL, Furlonger K, et al. Defective liver formation and liver cell apoptosis in mice lacking the stress signaling kinase SEK1/MKK4. Development 1999;126:505-516.
- Watanabe T, Nakagawa K, Ohata S, Kitagawa D, Nishitai G, Seo J, Tanemura S, et al. SEK1/MKK4-mediated SAPK/JNK signaling participates in embryonic hepatoblast proliferation via a pathway different from NF-kappaB-induced anti-apoptosis. Dev Biol 2002;250:332-347.
- Wada T, Joza N, Cheng HY, Sasaki T, Kozieradzki I, Bachmaier K, Katada T, et al. MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nat Cell Biol 2004;6:215-226.
- Wuestefeld T, Pesic M, Rudalska R, Dauch D, Longerich T, Kang TW, Yevsa T, et al. A Direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell 2013;153:389-401.
- Ooshio T, Yamamoto M, Fujii K, Xin B, Watanabe K, Goto M, Okada Y, Suzuki A, Penninger J, Nishina H, and Nishikawa Y. Hepatocyte MKK7 Contributes to Restoration of the Liver Parenchyma Following Injury. Hepatology 2020; doi: 10.1002/hep.31565. Online ahead of print.
- Pohl JF, Melin-Aldana H, Sabla G, Degen JL, Bezerra JA. Plasminogen deficiency leads to impaired lobular reorganization and matrix accumulation after chronic liver injury. Am J Pathol 2001;159:2179-2186.
- Nagahama Y, Sone M, Chen X, Okada Y, Yamamoto M, Xin B, Matsuo Y, et al. Contributions of hepatocytes and bile ductular cells in ductular reactions and remodeling of the biliary system after chronic liver injury. Am J Pathol 2014;184:3001-3012.
- Yu H, Konigshoff M, Jayachandran A, Handley D, Seeger W, Kaminski N, Eickelberg O. Transgelin is a direct target of TGF-beta/Smad3-dependent epithelial cell migration in lung fibrosis. Faseb J 2008;22:1778-1789.
- McNeill H, Jensen PJ. A high-affinity receptor for urokinase plasminogen activator on human keratinocytes: characterization and potential modulation during migration. Cell Regul 1990;1:843-852.
- Huang S, Liu F, Niu Q, Li Y, Liu C, Zhang L, Ni D, et al. GLIPR-2 overexpression in HK-2 cells promotes cell EMT and migration through ERK1/2 activation. PLoS One 2013;8:e58574.
- Heckel JL, Sandgren EP, Degen JL, Palmiter RD, Brinster RL. Neonatal bleeding in transgenic mice expressing urokinase-type plasminogen activator. Cell 1990;62:447-456.
- Currier AR, Sabla G, Locaputo S, Melin-Aldana H, Degen JL, Bezerra JA. Plasminogen directs the pleiotropic effects of uPA in liver injury and repair. Am J Physiol Gastrointest Liver Physiol 2003;284:G508-515.
- Bezerra JA, Bugge TH, Melin-Aldana H, Sabla G, Kombrinck KW, Witte DP, Degen JL. Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver. Proc Natl Acad Sci U S A 1999;96:15143-15148.
- Bezerra JA, Currier AR, Melin-Aldana H, Sabla G, Bugge TH, Kombrinck KW, Degen JL. Plasminogen activators direct reorganization of the liver lobule after acute injury. Am J Pathol 2001;158:921-929.
- Murakami T, Yasuda Y, Mita S, Maeda S, Shimada K, Fujimoto T, Araki S. Prealbumin gene expression during mouse development studied by in situ hybridization. Cell Differ 1987;22:1-9.
- Weisend CM, Kundert JA, Suvorova ES, Prigge JR, Schmidt EE. Cre activity in fetal albCre mouse hepatocytes: Utility for developmental studies. Genesis 2009;47:789-792.

