





研究交流
核内受容体CAR及びPXRと肝化学発がん
吉成浩一
静岡県立大学薬学部衛生分子毒性学分野
1. はじめに
核内受容体CAR及びPXRは、肝に高発現し、生体外異物の結合により活性化して薬物代謝酵素や薬物トランスポーターの遺伝子発現を調節する。両受容体の標的遺伝子はほぼ共通しており、代表的なヒト遺伝子としてCYP3A4やCYP2B6などが知られている。一方、両受容体の活性化物質には差が認められ、代表的なCAR活性化物質としては抗てんかん薬のフェノバルビタール(PB)が、ヒトPXR活性化物質としては抗結核薬のリファンピシンが知られている。
CAR活性化薬のPBは肝発がんプロモーターであることから、CARと肝化学発がんの研究が進められ、PBの肝発がんプロモーション作用にはCARが必須であることが証明された[1]。しかし、CAR依存的な肝発がんの分子機序は未だに明確にはなっていない[2,3]。また、CARとPXRは機能的に類似しているが、肝化学発がんに対するPXRの寄与は全く不明であった[2,3]。本稿では、CAR及びPXRと肝細胞増殖・肝発がんに関する最近の著者らの研究成果を紹介する。
2. CARと肝細胞増殖・肝発がん
マウス肝において、CARの活性化に伴い、肝細胞増殖の誘導、MYCとその標的遺伝子Foxm1の発現誘導、また、p53やGADD45B、サイクリンD1の発現亢進などが起こることが報告されているが、CAR活性化とこれら因子の因果関係は不明である[3,4]。また、PBの肝発がんプロモーション作用は齧歯動物特異的であり、ヒトでは認められないとされている [5]。一方、PBによるCARの活性化と薬物代謝酵素遺伝子の発現誘導には種差が認められず、また、CARのDNA結合領域は種間で高度に保存されているのに対して、リガンド結合領域(LBD)には大きな種差が認められる[3]。これらのことから筆者らは、CARはDNA結合ではなく、LBDを介したタンパク質間相互作用により肝細胞増殖を引き起こす可能性を考えた。さらに、CARの活性化は肝肥大を起こすことから、臓器サイズを決定するHippoパスウェイ[6-8] に着目した。Hippoパスウェイは肝サイズを規定する一方で、そのエフェクター分子であるYAPの異常な活性化は肝がんを誘発し、多くの肝がん組織ではYAPの活性化(核内蓄積)が起こる[7-10]。さらに、マウスにCARリガンドのTCPOBOPを投与すると、肝細胞増殖とともにYAPの核内蓄積が起こることも報告されていた [4]。そこで著者らは、CAR依存的な肝発がんにはYAPの活性化が関与するとの作業仮説を立て、その検証を進めた[11]。
TCPOBOPの投与によりCAR標的遺伝子Cyp2b10と共に細胞周期関連遺伝子の発現も亢進した。そして、この条件下では、YAPは核内に蓄積し、YAP標的遺伝子(Birc5、Ankrd1、Myc)の発現亢進も確認された(図1)。また、YAP/TEAD阻害薬のverteporfinとTCPOBOPの共処置により、TCPOBOP依存的な肝肥大は抑制され、Ankrd1やMacm2の発現も抑制傾向を示した[11]。
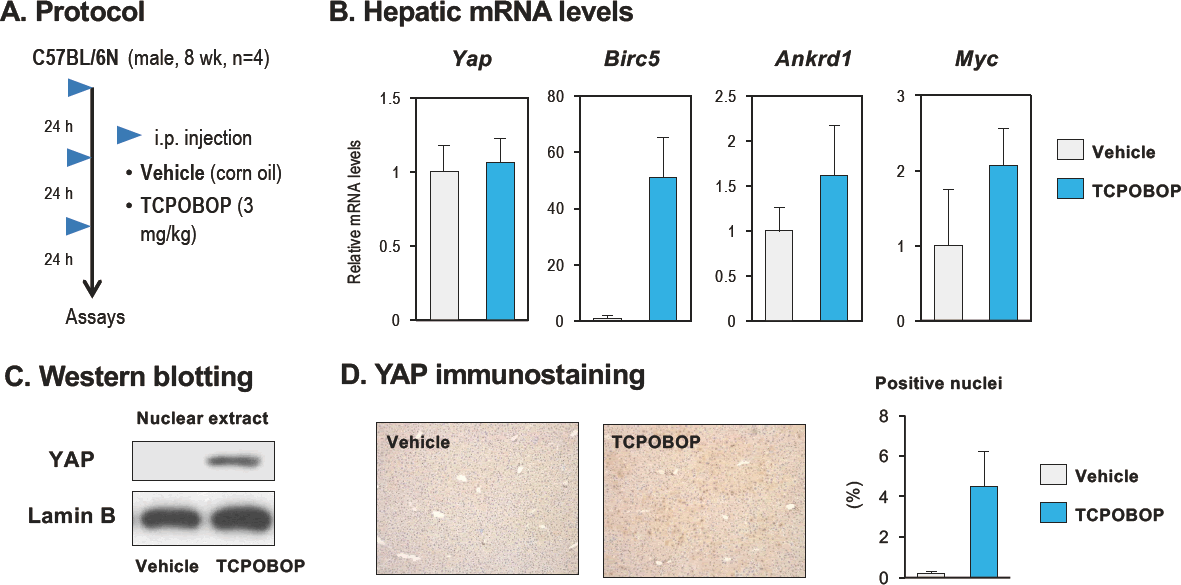
図1.CAR活性化に伴うYAPの活性化
A:マウスにCARリガンドTCPOBOP又は溶媒を腹腔内投与した。
B:定量的逆転写PCR法によりYAP標的遺伝子の肝mRNAレベルを測定した。
C:肝各抽出液を用いたウェスタンブロットにより核内YAPレベルを測定した。
D:抗YAP抗体を用いて肝組織を染色した。(B、C:文献11よりデータを引用して作図)
肝細胞におけるCARの発現は初代培養に移すと急激に低下し、培養肝細胞ではCAR依存的な遺伝子発現応答は劇的に減弱することから、初代培養肝細胞及びマウス肝細胞株AML-12細胞にアデノウイルスを用いてマウスCARを強制発現したところ、CAR標的遺伝子の発現亢進、CAR依存的な細胞増殖並びにYAP標的遺伝子やサイクリン類の発現亢進が認められた[11]。そこで、この試験系においてsiRNAを用いたYAPのノックダウン並びにverteporfin処置を行ったところ、CAR依存的な細胞増殖並びにYAP標的遺伝子や細胞増殖関連遺伝子の発現誘導はほぼ消失した(図2)。興味深いことにYAPのノックダウンやverteporfin処置は、血清刺激によるAML-12細胞の増殖には影響しなかった[11]。
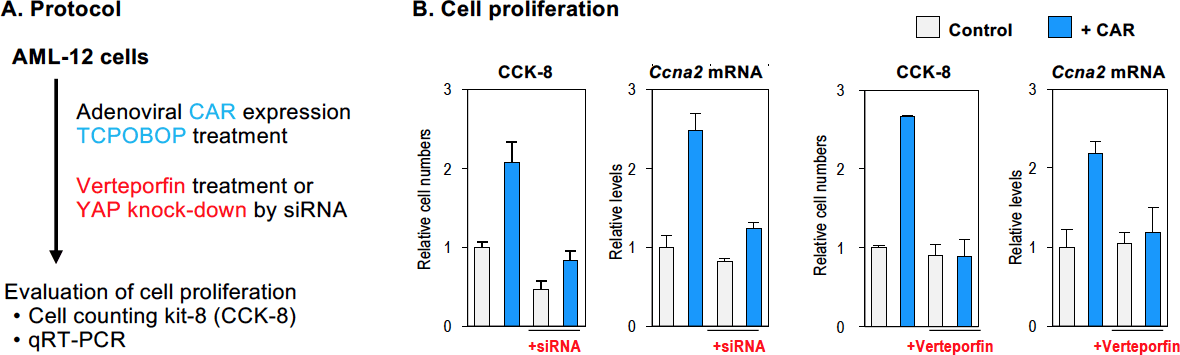
図2.マウス肝細胞のCAR依存的な増殖に対するYAPの寄与
アデノウイルスベクターを利用してAML-12細胞にマウスCARを発現し、CARリガンドTCPOBOP又は溶媒を処置した。その後、verteporfin処置又はYAPのsiRNAを導入し、細胞数及びCcna2のmRNAレベルを測定した。(B:文献11よりデータを引用して作図)
以上の結果から、CAR依存的なマウス肝細胞の増殖にはYAPの活性化が必要であることが強く示唆された。CARの活性化は、薬物代謝酵素誘導により活性酸素種の生成を引起こす一方で、YAPを活性化して肝細胞増殖の促進やアポトーシスの抑制を起こし、これらの相乗的作用により、PBは肝がんを誘発すると考えられる。筆者らは、CARはそのLBDを介してYAPと相互作用することを見出しているが(投稿準備中)、現時点ではCARの活性化がどのような機序でYAPの活性化を引き起こすかは不明である。正常肝細胞ではCARとYAPはともに細胞質に存在し、活性化により核内に移行することから、細胞内局在の調節機構に着目し、研究を進めている。

図3.CAR依存的な肝発がんにおけるYAPの役割
3. PXRの肝細胞増殖増強作用
PXRは、薬物代謝酵素の発現調節ではCARと類似した作用を示すが、肝細胞増殖・肝発がんに関しては非常に異なる性質を示し、PXRの活性化は肝細胞増殖を誘発しない。PXRの活性化により肝細胞肥大や肝肥大が起こるが、これは酵素誘導に伴うものであると考えられている[12]。しかし、高用量、長期間のPXRリガンド(pregnenolone 16α-carbonitrile; PCN)の投与でマウス肝のPCNA陽性細胞数が増加すること、大腸がん細胞や神経芽腫細胞にPXRを高発現すると増殖が抑制されること、PXR欠損マウスでは部分肝切除後の回復が遅れることなども報告されている[13-15]。そこで筆者らは、肝細胞増殖・肝発がんとPXRの関連について研究を開始した[16,17]。
マウスにPXRリガンドのPCNとCARリガンドのTCPOBOPを投与して肝細胞増殖を評価した。このとき、PXRとCARは類似した標的遺伝子を有すること、PXRの活性化は細胞増殖を抑制するという報告があったことから[15]、PXRがCAR依存的な肝細胞増殖を抑制する可能性を考え、併用投与も行った。その結果、TCPOBOP投与は非常に強く肝細胞増殖を誘導したのに対して、PCN単独投与は細胞増殖マーカーに全く影響を与えなかった(図4)。一方、併用投与群では、TCPOBOP単独投与群に比べて、顕著な肝細胞増殖の誘導が認められた(図4)。これらのPCN処置の効果は、PXR欠損マウスでは認められなかった[16]。興味深いことに、薬物代謝酵素遺伝子(Cyp2b10)発現誘導は、TCPOBOP単独投与群と併用投与群で同程度であった(図4)。さらに、PCN投与は、CARと同様に齧歯動物において肝細胞増殖を引き起こす核内受容体PPARαを介した肝細胞増殖に対しても増強作用を示したが、代謝酵素遺伝子(Cyp4a10)の発現誘導には影響を与えなかった[16]。また、当研究室においてマウスPXR活性化物質として見出した防かび剤イマザリルもまたTCPOBOP依存的な肝細胞増殖を増強した[18]。これらの結果から、PXRの活性化はCAR依存的な肝細胞増殖を増強し、これは単にCARの転写因子としての機能を増強するのではなく、間接的な機序によると考えられた。

図4.PXRによるCAR依存的肝細胞増殖の増強
雄性C56BL/6マウスにCARリガンドTCPOBOP(TC)とPXRリガンドPCNを単独又は併用投与し、48時間後に屠殺した。
A: 抗Ki-67抗体を用いた肝組織切片の免疫組織染色を行い、陽性核数をカウントした。
B:定量的逆転写PCR法により肝mRNAレベルを測定した。(文献16よりデータを引用して作図)
次に、四塩化炭素(CCl4)誘発急性肝障害に伴う肝再生時の肝細胞増殖に対するPXR活性化の影響を解析した(図5)。マウスにCCl4を投与し、24時間後にPCNを単回投与して経時的に肝細胞増殖の程度を評価したところ、Ki-67陽性細胞数や細胞周期関連遺伝子の発現は、CCl4単独投与時ではCCl4投与72時間をピークとする変動パターンを示したのに対し、PCN併用投与群ではそのピークはより早期に移行した。さらに、CCl4単独投与48時間後ではほとんど認められないM期肝細胞の数がPCNの併用処置により著しく増加した。さらに、マウス個体における組換えFGF19投与依存的な肝細胞増殖、並びにAML-12細胞における血清依存的な増殖に対しても、PXRの活性化は増強作用を示した[17]。以上の結果から、PXRは、化学物質だけでなく、増殖因子による肝細胞増殖も増強することが明らかとなった。
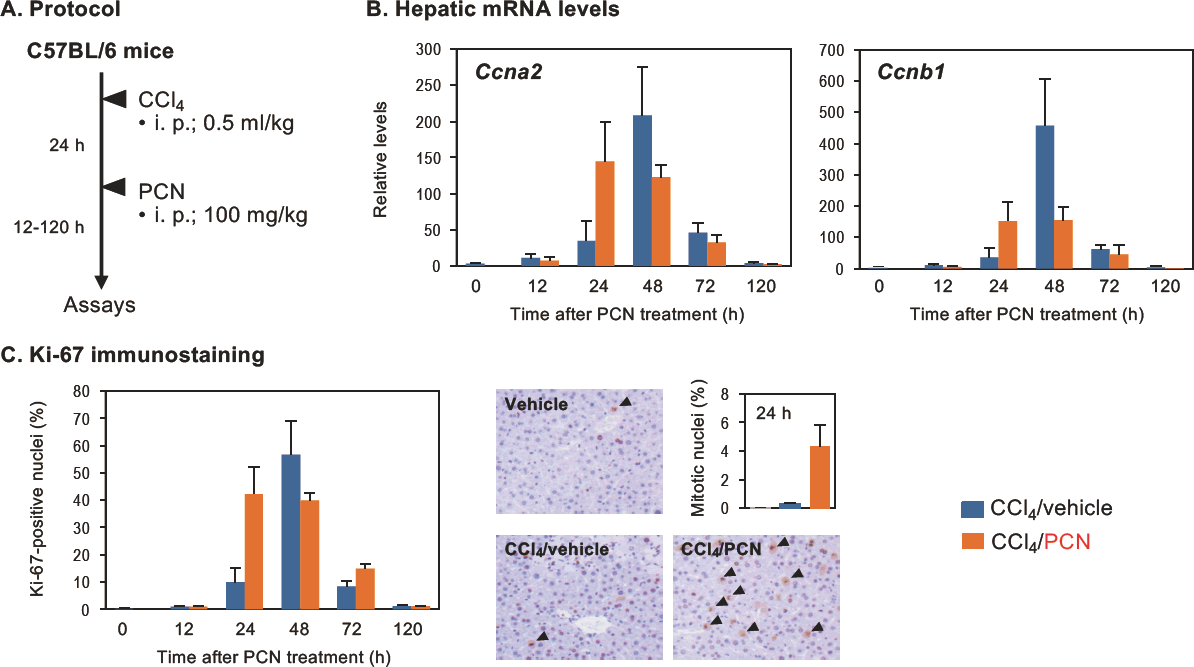
図5.PXRによる肝障害依存的肝細胞増殖の増強
A:雄性マウスにCCl4を投与し、24時間後にPCN又は溶媒(コーン油)を投与して経時的に屠殺した。
B:定量的逆転写PCR法により肝mRNAレベルを測定した。
C:抗Ki-67抗体を用いた肝組織切片の免疫組織染色を行い、陽性核数(左)並びにM期細胞数(右、PCN投与24時間後)をカウントした。(B、C:文献17よりデータを引用して作図)
PXRの肝細胞増殖増強作用は、増殖刺激の種類に依らないことが示唆されたため、PXR活性化は細胞周期調節機構そのものに影響を及ぼすのではないかと仮説を立て、解析を進めた[17]。その結果、PCN投与により、マウス肝におけるCdkn1b(p27)やRbl2(p130)、Cdkn1a(p21)等の細胞周期抑制性因子の発現が有意に低下すること、この低下はPXR欠損マウスでは認められないことが明らかとなった。さらに、PXRは、これら細胞周期抑制因子の遺伝子発現を正に制御するFOXO3と相互作用し、FOXO3依存的な遺伝子発現を抑制することで、これら因子の発現低下を引き起こすことが示唆された。
以上の結果から、PXRの活性化は、細胞周期抑制因子の発現低下を介して肝細胞の細胞増殖刺激に対する感受性を亢進し、様々な細胞増殖刺激に対して増強作用を示すと考えられた(図6)。
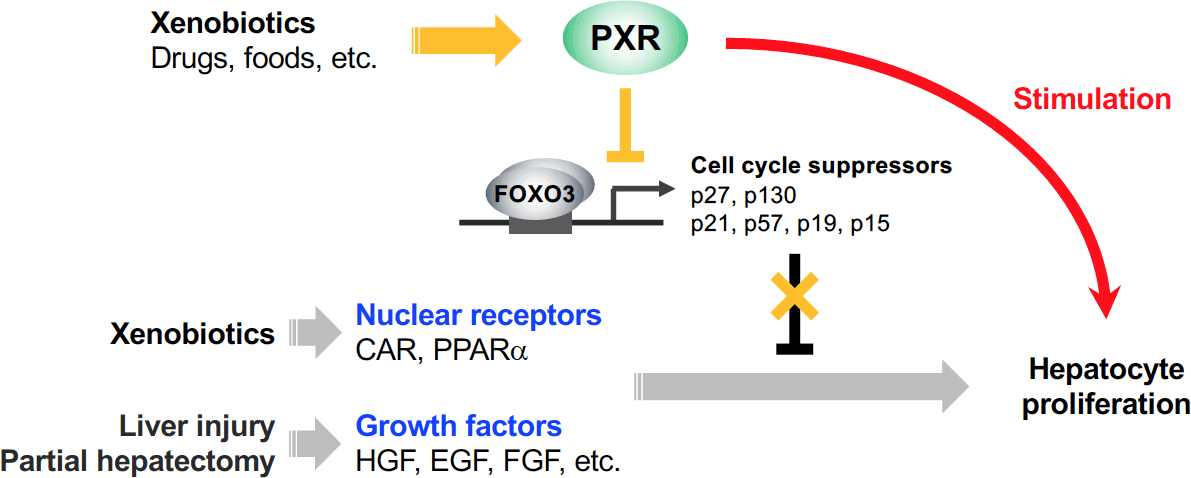
図6.PXR活性化に伴う肝細胞増殖増強の推定分子機序
PXRは、FOXOファミリー転写因子依存的な細胞周期抑制因子の発現を抑制することで様々な肝細胞増殖刺激に対する感受性を亢進すると考えられた。
4. PXRと肝発がん
上記の結果から、PXR活性化物質は、発がん作用を示さない一方で、肝発がん物質に対する感受性を亢進させる発がん促進因子として働く可能性が考えられた。そこで、多段階肝発がんモデルを利用して、PXRの肝発がん作用並びにCAR依存的肝発がんに対する修飾作用を解析した[19]。
発がんイニシエーターのジエチルニトロサミン投与後、PBとPCNを単独又は併用投与した。投与開始20週間後に一部のマウスを屠殺して肝臓の前がん性変化をケラチン8/18(KRT8/18)の免疫組織染色により評価したところ、PCN単独投与群ではKRT8/18陽性細胞はほとんど認められなかったが、PB単独投与群では著しいKRT8/18陽性細胞数の増加が認められ、PCNの併用により増加はより顕著となった[20]。残りのマウスをPB/PCN投与開始35週間後に屠殺し、腫瘍のサイズと発生頻度を評価した(図7)。その結果、PB単独投与群又はPB/PCN併用投与群では全マウスで癌腫又は好酸性腺腫の発生が認められたが、癌腫のサイズ(平均直径)、癌腫/好酸性腺腫の個体あたりの発生数、並びPCNA陽性細胞面積は、PB単独投与群に比べて併用投与群で顕著に減少していた。また、PCN単独投与では有意な腫瘍形成は認められなかった。
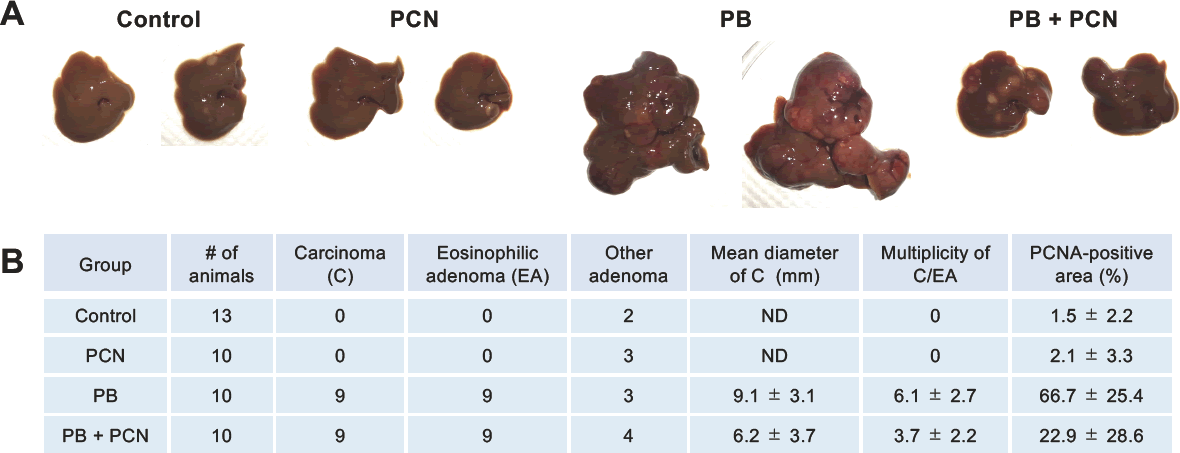
図7.二段階発がんモデルにおけるPXRの発がん及び発がん修飾作用の評価
雄性C3Hマウスにジエチルニトロサミン(90 mg/kg)を腹腔内投与し、2週間後よりPB(飲水、500 ppm)とPCN(混餌、100 ppm)を単独又は併用投与した。投与開始35週間後に屠殺し、肝腫瘍の発生を肉眼的(A)及び組織染色(ヘマトキシリン・エオシン)標本の顕微鏡観察(B)により評価した。また、抗PCNA抗体を用いた肝組織切片の免疫組織染色を行い、陽性面積を測定した(B)。(B:文献19よりデータを引用して作図)
以上の結果から、PXR活性化物質は少なくてもマウスにおいては肝発がん作用を示さないと考えられた。また、PXRの活性化は、PBによる肝細胞増殖や前がん性変化を促進するが、肝腫瘍の進展や悪性化を抑制することが示唆された。したがって、PXRは肝発がんの異なる段階において促進と抑制の両面性の作用を示す可能性が考えられた。
5 おわりに
以上、筆者らは、異物応答性の核内受容体であるCAR及びPXRの肝発がんにおける役割とその作用機序に関する研究を進め、両受容体が薬物代謝の制御では非常に類似した作用を示すのに対し、肝細胞増殖や肝発がんに関しては全く異なる作用を示すことを明らかにした[2,3,11,16-19]。特に、PXRの作用は非常にユニークであり、発がんモデルを用いた解析結果はPXR活性化物質が肝がん抑制物質として働く可能性を示していることから、抗がん薬としての創薬応用を見据え、その作用機序に関する研究を進めている。他方、CARを介した肝発がんに関してYAPの関与を示唆する結果が得られたが、CARがどのようにYAPの活性化を制御しているのかについては分かっていない。代表的な肝発がんプロモーターであるPBの発がん機序の全容解明は、化学発がん研究における大きな課題の1つであり、その解決に向けてCARとYAPのクロストークの分子機序解明を進めている。
謝辞
本稿で紹介した研究は、筆者の前任地の東北大学大学院薬学研究科薬物動態学分野及び現所属で実施したものです。本研究の遂行に中心的に携わった阿部太紀博士(現東北大学大学院医学系研究科)と志津怜太博士(静岡県立大学薬学部)、並びに本研究に携わった全ての方に感謝いたします。また、本研究会の世話人にご推薦いただいた塩尻信義先生(静岡大学)に感謝いたします。最後に、このような研究紹介の機会をご提供いただいた本研究会の諸先生に御礼申し上げます。
参考文献
- Y. Yamamoto, et al., The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res. 64:7197-7200, 2004.
- K. Yoshinari, Role of nuclear receptors PXR and CAR in xenobiotic-induced hepatocyte proliferation and chemical carcinogenesis. Biol Pharm Bull. 42: 1243-1252, 2019.
- R. Shizu, K. Yoshinari, Nuclear receptor CAR-mediated liver cancer and its species differences. Expert Opin Drug Metab Toxicol. 16: 343-351, 2020.
- M. A. Kowalik, et al., Yes-associated protein regulation of adaptive liver enlargement and hepatocellular carcinoma development in mice. Hepatology. 53: 2086-2096, 2011.
- International Agency for Research on Cancer (IARC). Some thyro- tropic agents, IARC monographs on the evaluation of carcinogenic risk to humans. Vol. 79, pp. 161–288 (2001).
- G. Halder, R.L. Johnson, Hippo signaling: growth control and beyond. Development. 138: 9-22, 2011.
- F. X. Yu, et al., Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 163: 811-828, 2015.
- G. K. Michalopoulos, Hepatostat: Liver regeneration and normal liver tissue maintenance. Hepatology. 65: 1384-1392, 2017.
- F. H. Kieran, The Hippo pathway and human cancer. Nat Rev Cancer. 13: 246-257, 2013.
- T. Zheng, et al., Hippo signaling in oval cells and hepatocarcinogenesis. Cancer Lett. 302: 91-99, 2011.
- T. Abe, et al., Role of YAP activation in nuclear receptor CAR-mediated proliferation of mouse hepatocytes. Toxicol Sci. 165: 408-419, 2018.
- R. R. Maronpot, et al., Hepatic enzyme induction: histopathology. Toxicol Pathol. 38: 776-795, 2010.
- J. Staudinger, et al., Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos, 29: 1467-1472, 2001.
- G. Dai, et al., Pregnane X receptor is essential for normal progression of liver regeneration. Hepatology. 47: 1277-1287, 2008.
- N. Ouyang, et al., Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells. Br J Cancer. 102: 1753–1761, 2010.
- R. Shizu, et al., Xenobiotic-induced hepatocyte proliferation associated with constitutive active/androstane receptor (CAR) or peroxisome proliferator-activated receptor α (PPARα) is enhanced by pregnane X receptor (PXR) activation in mice. PLoS One. 8: e61802, 2013.
- R. Shizu, et al., PXR stimulates growth factor-mediated hepatocyte proliferation by cross-talk with FOXO transcription factor. Biochem J. 473: 257-266, 2016.
- S. Yoshimaru, et al., Acceleration of murine hepatocyte proliferation by imazalil through the activation of nuclear receptor PXR. J Toxicol Sci. 43: 443–450, 2018.
- R. Shizu, et al., The influence of the long-term chemical activation of the nuclear receptor pregnane X receptor (PXR) on liver carcinogenesis in mice. Arch Toxicol. 95: 1089-1102, 2021.

