





研究交流
ヒトiPS細胞を利用した肝疾患モデル
東京医科歯科大学 大学院医歯学総合研究科 疾患生理機能解析学分野
柿沼 晴
はじめに
ヒト体細胞を人為的にリプログラミングすることで、体を構成する全ての細胞に誘導が可能となる人工多能性細胞(iPS細胞)を樹立できることが、山中らによって報告された[1]。ES細胞やiPS細胞から誘導された肝細胞様細胞は、再生医学的治療の開発、あるいは創薬研究などに用いられる一方で、肝臓を体外培養器官として模倣する「肝臓オルガノイド」を作成することが可能であることも報告されている[2]。現時点では、臓器としての肝臓をiPS細胞から創り出し、模倣するには、いくつかのハードルが残されているが、単一あるいは2-3種程度の細胞種からなる組織様培養体を作って疾患モデルとすること、あるいは細胞間相互作用の解析ツールとしては非常に有用な存在であることが示されつつある。我々はこれまで、ヒトiPS細胞を利用したいくつかの肝疾患モデルを樹立し、報告してきた[3-5]。本稿では、最近報告したいくつかの肝線維化に関わる疾患モデル研究について取り上げ、ヒトiPS細胞を利用した肝疾患モデル研究について概説したい。
先天性肝線維症(Congenital Hepatic Fibrosis: CHF)
CHFは、PKHD1(Polycystic Kidney and Hepatic Disease 1)を責任遺伝子とする遺伝性肝疾患であり、進行性の肝線維化と細胆管の増生とを特徴とする。進行例では、小児期に肝不全を呈して肝移植が必要となる場合もある。本症では、炎症細胞の浸潤が乏しく、肝星細胞の活性化が乏しいこと、門脈域周囲にのみ広範な線維化が生じることから、通常の慢性肝炎に伴う肝硬変とは異なる線維化の病態があると考えられる[6]。本症は、胎生期の胆管原器に異常を生じるductal plate malformationに起因することが示唆されているが、その病態には多くの不明点が残されている。そこで我々は、本症を模倣したiPS細胞培養系を構築することで、疾患モデルとして標的分子を探索できないかと考えた。すなわち、疾患型ヒトiPS細胞株から胆管細胞系譜へ分化誘導し、本症で見られる特異な肝線維化のメカニズムを解明することを試みた。
本症の責任遺伝子であるPKHD1はゲノム全長 470,000 bp、本遺伝子がコードする蛋白であるFibrocystinは440 kDaからなる巨大な分子である。Fibrocystin蛋白は、胆管上皮においては、管腔側に伸びて機械的・化学的刺激を受容する一次繊毛の基部に局在することが知られている[7]。従って、本症では胆管上皮の一次繊毛に機能的異常があると推測されるが、それが何故、前記のようなductal plate malformationと肝星細胞の活性化が乏しい肝線維化とにつながるのかが不明である。iPS細胞を用いた疾患病態研究では、患者由来iPS細胞を樹立する手法がよく用いられるが、コントロールとなる健常型細胞は当然他人に由来するもので、遺伝的背景が異なってしまううえに、iPS細胞そのものの未分化性、分化特性などにも差が出る可能性がある。そこで我々は、本疾患の研究には、患者由来のヒトiPS細胞を樹立するよりも、健常者由来ヒトiPS細胞株からゲノム編集によりPKHD1を改変してFibrocystin完全欠損型のiPS細胞を作製し、この表現型を解析することで、本症の病態に重要な分子機構を解明できるのではないかと考え、本研究を行った[3]。
iPS細胞による先天性肝線維症の疾患モデル
健常者由来ヒトiPS細胞株に対してCRISPR/Cas9法を用いて、完全欠損させたPKHD1-KO iPS細胞株、及び非特異的挿入変異を考慮したコントロールとしてヘテロ欠損株(PKHD1-Hetero iPS)を樹立した。これらのiPS細胞株を、既報[8]に従い、in vitro で肝芽細胞系譜に分化誘導して株化し、胆管細胞系譜への分化誘導を行い、その形質を解析した。 胆管細胞系譜への分化誘導により球形培養体が誘導された。これは細胞極性をもった一層のCK7陽性細胞から構成され、胆管細胞関連分子の発現が亢進していることから、胆管細胞の形質を呈するcholangiocytic cysts (CCs)であることが示された(図1)。PKHD1-KO CCsではPKHD1遺伝子がコードするFibrocystinの発現が欠損しており、走査型電子顕微鏡による解析では、PKHD1-KO CCsでは一次繊毛の短縮と変形とが認められた。さらに、PKHD1-KO CCsでは、PKHD1-Hetero CCsに比べて大型のcystが有意に多く形成され、細胞増殖が亢進していることが明らかになった(図1)。
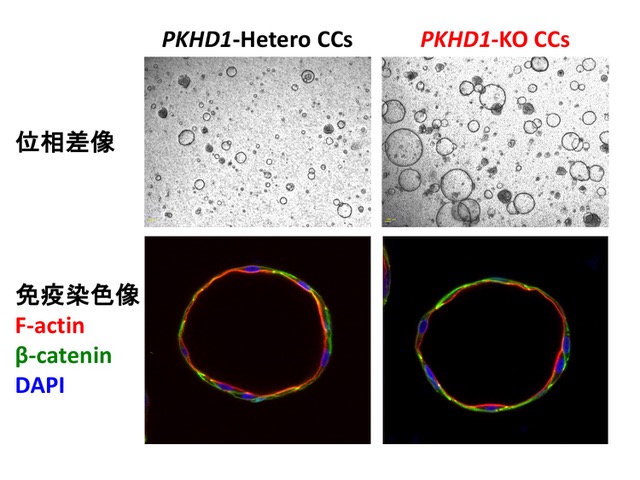
図1:先天性肝線維症モデルとしてのヒトiPS細胞由来胆管細胞の誘導
先天性肝線維症の責任遺伝子であるPKHD1(Fibrocystin)について、完全欠損させたPKHD1-KO iPS細胞株、及び非特異的挿入変異を考慮したコントロールとしてヘテロ欠損株(PKHD1-Hetero iPS)を樹立した。iPS細胞から肝芽細胞系譜へ分化誘導した後に、胆管細胞系譜に誘導しつつ、管腔構造を模倣する培養体(Cholangiocytic cysts: CCs)を形成させた。Fibrocystinを欠損するiPS細胞から誘導した場合、より大きく多数の胆管構造が誘導され、これらでは細胞増殖の異常な亢進がみられた(文献3より改変して引用)。
網羅的発現解析により変動因子をみると、PKHD1-KO CCsにおいてインターロイキン(IL)-8の発現が顕著に亢進していた。IL-8産生は蛋白レベルでもPKHD1-KO CCで有意に亢進していることを確認したため、IL-8の関与を検証したところ、PKHD1-KO CCsに対してIL-8受容体拮抗剤を添加すると細胞増殖が抑制され、逆にPKHD1-Hetero CCsではIL-8の添加により細胞増殖が促進した。このことから、CHF型胆管細胞では、IL-8依存的にCCsの細胞増殖亢進がみられることが明確に示された。
次に、線維化に関与する分子を解析すると、PKHD1-KO CCsでは対照と比較してCTGF(結合組織成長因子)の産生が亢進していた。PKHD1-KO CCsにIL-8受容体拮抗剤を添加するとCTGFの産生が抑制され、Hetero CCsへのIL-8添加ではCTGFの産生上昇が認められたことから、PKHD1-KO CCs におけるCTGF産生もIL-8依存的であることが示された。
IL-8およびCTGFの産生がPKHD1-KO CCsで亢進する機序を検討したところ、PKHD1-KO CCsではHetero CCsに比べてERKを含めたMAPキナーゼ経路の活性化が認められた。PKHD1-KO CCsに対して選択的MEK/ERK経路阻害剤を添加すると、IL-8およびCTGFの産生とCC形成が有意に抑制された。これらのことから、PKHD1-KO CCsでは自律的なERK経路の活性化がIL-8やCTGFの産生亢進を誘導することが示された。
最後に検証解析として、先天性肝線維症症例の血清及び肝生検検体を用いて解析を行った。CHF症例の血清中IL-8は全例で対照群(同年齢小児のC型慢性肝炎症例)に比して高値であった。定量的PCRを用いた肝組織中の遺伝子発現比較では、CHF症例において肝組織中のIL-8およびCTGFの発現が、C型慢性肝炎例と比較して有意に亢進していた[3]。
これらの結果から、CHFにおいてはIL-8およびCTGFの産生亢進が病態形成に重要であることが示された(図2)。さらに、IL-8シグナルの抑制により、異常な胆管形成と線維化の進展とを抑えられる可能性が示されたとも言える。このことは、ヒトiPS細胞培養系を用いた病態解析研究により、従来のマウスや臨床検体のみを用いた解析では指摘されていなかった、「IL-8が本症の重要な標的分子となる」ことを示した点でも意義が深いと考えられる。
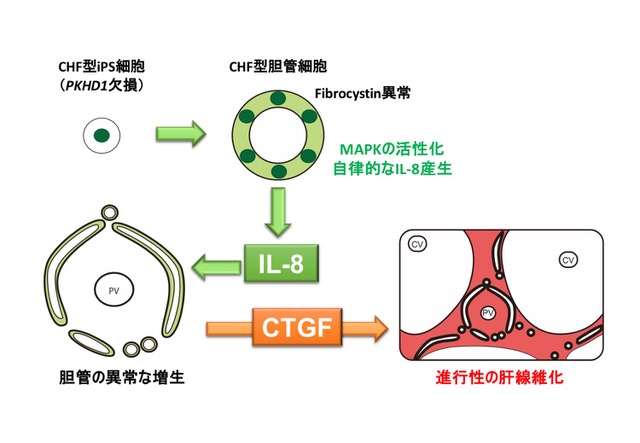
図2:ヒトiPS細胞を用いた疾患モデルにより明らかとなった先天性肝線維症の病態生理
先天性肝線維症では、先天的なFibrocystinの異常がある。 Fibrocystinの機能を失った胆管細胞では、IL-8の産生が自律的に異常に亢進してしまい、胆管細胞自身の過剰な増殖を引き起こすことで、異常な胆管構造が形成される。IL-8は、同時に胆管細胞からCTGF産生を亢進させることで、その周囲の線維芽細胞に作用して強い肝線維化を誘導する。
ヒトiPS細胞由来肝星細胞(iPS-HSCs)の誘導
ここまでは、遺伝性肝疾患に関して述べてきたが、未だ有効な薬物治療がない非アルコール性脂肪性肝炎などの慢性肝炎の線維化病態モデルとして、ヒトiPS細胞由来肝星細胞(iPS-HSCs)を誘導し利用することが期待され、報告されている[9-11]。我々はiPS-HSCsを誘導する独自の系の構築を試みた。ヒト肝星細胞の発生に関しては未だ不明な点が多く残されているが、マウスでの研究では一定の知見があり、中胚葉系譜、横隔間充織、中皮細胞を経て発生することが報告されている[12]。そこでヒトiPS細胞から中胚葉系譜への分化誘導に関する報告を基盤として、4つの因子(FGF-2、BMP-4、Activin A、GSK3β-inhibitor)を選択して誘導系を構築した[4]。誘導した細胞は肝星細胞に発現するALCAM、NGFR、HGF、PPARγといった遺伝子群を発現すること、ビタミンA貯蔵能があること、TGF-β1による活性化能を呈することから、肝星細胞と類似した形質を示すことが示され、既報[9-11]でみられるようなiPS由来肝星細胞 (iPS-HSCs)であると考え、以下の検討を行った。
転写因子LHX2はiPS-HSCsとiPS-HPCsとの細胞間相互作用を促進する
iPS-HSCsの機能的評価として、iPS-HPCsと共培養することで、iPS-HPCsの形質変化が誘導されるかについて検討した。iPS-HPCsとiPS-HSCsとを接触共培養したところ、iPS-HPCsにおけるアルブミン発現は、共培養により有意に顕著な発現亢進を認め、iPS-HPCsの肝成熟化を促進する効果を示した(図3・左)。
転写因子LHX2(LIM homeobox 2)は、マウスでは肝星細胞の活性化を抑制することが報告され、LHX2欠損マウスの肝臓では、胎生期から線維化が進展し、肝発育不全や造血障害により胎生致死に至る[13]。しかしながら、ヒト肝星細胞におけるLHX2の機能は未だ不明であり、今回誘導したiPS-HSCsを用いて、LHX2のヒト肝星細胞における機能解析を行った。
Doxycycline依存的にLHX2強制発現を誘導可能なヒトiPS細胞株(iLHX2)を作製した。このiLHX2から肝星細胞に誘導して作製したiLHX2-HSCsとiPS-HPCsとを接触共培養し解析を行ったところ、LHX2を強制発現したiLHX2-HSCsと共培養を行ったiPS-HPCsは、対照のDoxycycline非添加群に比べてα-fetoprotein、Albumin、アポ蛋白等の発現が有意に上昇しており、さらに培養上清中のAlbumin産生量も有意に増加し(図3・右)、肝細胞としての機能成熟化が促進されていることが示された。
次にLHX2発現増強iPS-HSCsによる機能成熟化促進のメカニズムについて解析した。非接触共培養系では、接触共培養で得られたLHX2の肝成熟化促進効果は認められなかったことから液性因子の関与は否定的だった。網羅的発現解析では、肝臓の細胞外マトリクスとして重要な構成成分であるコラーゲン、ラミニンの発現プロファイルがLHX2強制発現によって様々な変化をしていた。したがって、iPS-HSCsではLHX2発現を増強することで細胞外マトリクスの発現プロファイルが変化し、その変化がiPS-HPCsの肝細胞としての成熟化促進に重要であることが示唆され、iPS-HSCsにおけるLHX2の機能が本研究によって初めて示された[4]。
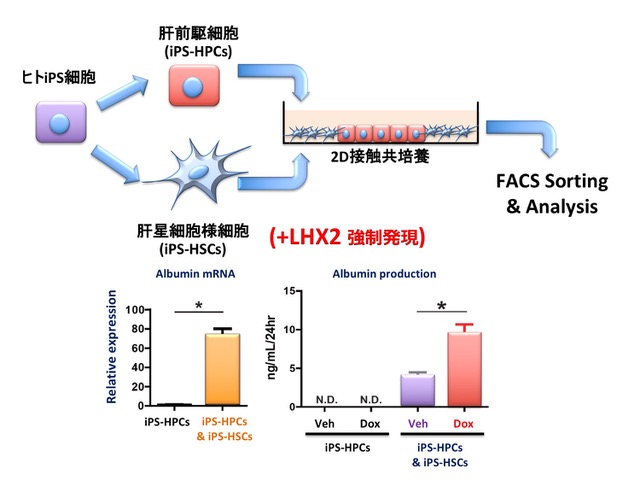
図3:ヒトiPS細胞由来肝星細胞様細胞(iPS-HSCs)と肝前駆細胞(iPS-HPCs)の細胞間相互作用の解析
iPS-HSCsとiPS-HPCsとの接触共培養を行うと、iPS-HPCsにおけるアルブミンの発現が50倍以上に増強される。さらに、LHX2をiPS-HSCsに強制発現すると、アルブミンの産生量は有意に増加する(文献4より改変して引用)。
おわりに
このように、ヒトiPS細胞から肝細胞系譜・胆管細胞系譜・肝星細胞系譜への分化誘導系を利用しながら細胞間相互作用や疾患のモデルを作製し、その病態生理を追求する研究を進めてきた。健常者由来のヒトiPS細胞から目的のゲノムのみを編集する手法、あるいは遺伝子異常や疾患特異的SNPを有する患者からiPS細胞を樹立する手法、の双方によって疾患モデルとなりうるiPS細胞を作製することが可能である。今後は、これを目的の細胞に誘導し、必要な細胞を選択して「肝オルガノイド」として細胞間相互作用や疾患病態モデルの解析をすすめてゆくことで、さらに新しい知見が開ける可能性がある(図4)。我々の進めている「疾患モデル」研究から、新しいバイオマーカーの発見・新しい治療法の開発へとつなげてゆきたいと考えている。
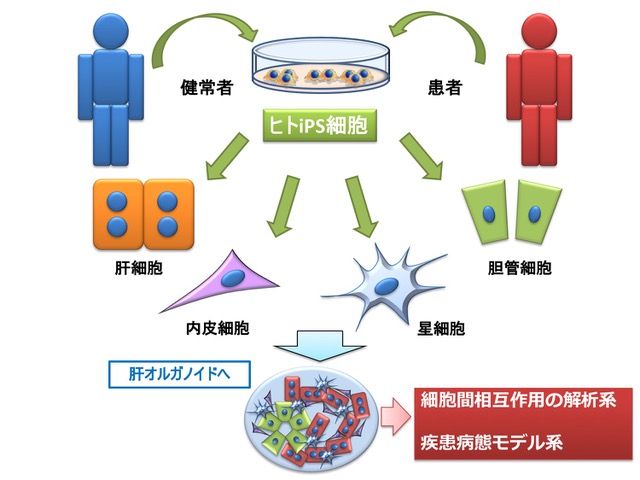
図4:ヒトiPS細胞由来肝構成細胞を用いた新たな肝オルガノイドの創生
健常者由来のヒトiPS細胞から標的ゲノムのみを編集する手法、あるいは遺伝子異常や疾患特異的SNPを有する患者からiPS細胞を樹立する手法、の双方によって疾患モデルとなりうるiPS細胞を作製することが可能である。ヒトiPS細胞は様々な肝構成細胞への誘導が可能となっている。必要な細胞を選択して「肝オルガノイド」として細胞間相互作用や疾患病態モデルの解析をすすめてゆくことで、さらに新しい知見が開ける可能性がある。
謝辞
最初に、2021年より本会の世話人に加えて頂きましたことを、大変光栄に存じますとともに関係の諸先生方に深謝申し上げます。微力ではございますが、本会の発展に少しでも貢献できるよう、さらに研究を進めて参りたいと存じます。
また、本稿で紹介した研究は東京医科歯科大学 消化器内科 渡辺 守 教授、朝比奈 靖浩 教授、三好 正人 博士、角田 知之 博士、東海大学医学部 紙谷 聡英 准教授、スタンフォード大学医学部 中内 啓光 教授らとの共同研究であり、論文共著者の先生方に深く感謝を申し上げます。
【引用文献】
- Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861-72.
- Ouchi R, Togo S, Kimura M. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab 2019;30:374-384.
- Tsunoda T, Kakinuma S, Miyoshi M, et al. Loss of fibrocystin promotes interleukin-8-dependent proliferation and CTGF production of biliary epithelium. J Hepatol 2019;71:143-152.
- Miyoshi M, Kakinuma S, Kamiya A, et al. LIM homeobox 2 promotes interaction between human iPS-derived hepatic progenitors and iPS-derived hepatic stellate-like cells. Sci Rep 2019;9:2072.
- Kaneko S, Kakinuma S, Asahina Y, et al. Human induced pluripotent stem cell-derived hepatic cell lines as a new model for host interaction with hepatitis B virus. Sci Rep 2016;6:29358.
- Nakanuma Y, Harada K, Sato Y, Ikeda H. Recent progress in the etiopathogenesis of pediatric biliary disease, particularly Caroli's disease with congenital hepatic fibrosis and biliary atresia. Histol Histopathol 2010;25:223-35.
- Zhang MZ, Mai W, Li C, et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci USA 2004;101:2311-6.
- Yanagida A, Ito K, Chikada H, et al. An in vitro expansion system for generation of human iPS cell-derived hepatic progenitor-like cells exhibiting a bipotent differentiation potential. PLoS One 2013;8:e67541.
- Koui Y, Kido T, Ito T, et al. An in vitro human liver model by iPSC-derived parenchymal and non-parenchymal cells. Stem Cell Reports 2017;9:490-498.
- Camp JG, Sekine K, Gerber T, et al. Multilineage communication regulates human liver bud development from pluripotency. Nature 2017;546:533-538.
- Coll M, Perea L, Boon R, et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 2018;23:101-113 e7.
- Asahina K, Tsai SY, Li P, et al. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 2009;49:998-1011.
- Wandzioch E, Kolterud A, Jacobsson M, et al. Lhx2-/- mice develop liver fibrosis. Proc Natl Acad Sci USA 2004;101:16549-54.

