





研究交流
肝細胞性腫瘍の表現型決定メカニズム:癌遺伝子シグナリング経路の相互作用による脱分化と分化転換
西川祐司
旭川医科大学病理学講座腫瘍病理分野
はじめに
肝に発生する腫瘍の大部分を占める上皮性悪性腫瘍は、肝細胞癌(hepatocellular carcinoma, HCC)と肝内胆管癌(cholangiocarcinoma, CC)に代表されるが、両者がさまざまな割合で混在 するあるいは 両者の中間的な形態をとる混合型肝癌(combined hepatocellular and cholangiocarcinoma, cHCC-CC)が存在する[1] 。これまでは形態学的な特徴に基づき、肝細胞癌は肝細胞に、胆管癌は胆管上皮細胞に由来すると考えられてきた。一方、混合型肝癌の 一部は、成熟肝に少数存在して 肝細胞と胆管上皮細胞の両方に分化可能な 肝幹細胞に由来する腫瘍と想定され、これはWHO 消化器腫瘍分類(第4版、2010年)にも反映されている。現在の腫瘍の病理学的分類の枠組みは、正常細胞の表現型との類似性から腫瘍の細胞起源を推定することで確立されてきたものである。しかし、腫瘍化に伴って生じうる種々の細胞分化異常を考慮すると、腫瘍細胞の表現型から細胞起源を求める方法が常に正しい結論を導き出すとは限らない。
成熟肝における上皮系細胞、すなわち肝細胞と胆管上皮細胞は著しく異なる形態学的・生理学的特徴を示しているが、これらは発生学的に共通の肝芽細胞に由来している[2]。肝細胞はその高次機能のゆえに、最終分化するとその表現型が固定されると長く信じられてきたが、我々は、成熟肝細胞は胆管上皮細胞へ分化転換する能力を保持していることを報告してきた[3-5] 。さらに、肝傷害に伴い、いったん成熟した肝細胞が幼若化(脱分化)し、肝幹細胞の性質を獲得する可能性も示唆されている[6,7] 。2012年に肝内胆管癌が肝細胞から発生しうることが実験的に証明され[8] 、その後、我々を含めた多くのグループにより肝上皮系腫瘍の細胞起源が再検討されている。肝上皮系腫瘍の成り立ちを理解する上で、分化転換、脱分化を含めた肝細胞分化異常の関与を明らかにすることは重要である。
我々はマウス肝腫瘍モデルの検討から、肝細胞は腫瘍化に伴い、胎児肝で発現する遺伝子群を再び発現することを明らかにしてきた[9] 。また我々は、種々の癌遺伝子をトランスポゾンシステムで肝細胞ゲノムに導入し、短期間に肝腫瘍を誘導する系を用いて、癌遺伝子の組み合わせと腫瘍表現型の関連性を検討してきた[10-12] 。本稿では我々のこれまでの実験結果を紹介し、成熟肝細胞に由来する腫瘍が多彩な組織像を示しうること、さらにこれらの表現型が種々の癌遺伝子の相互作用による分化状態の変化により規定されていることを述べたい。
マウス肝腫瘍における脱分化:胎児期・新生児期遺伝子発現の再活性化
ヒト肝細胞癌は慢性肝炎や肝硬変を基盤として発生することが多いが、明らかな異常のない肝から発生することもあり、肝細胞癌の発生過程は一様ではないことが示唆される。マウスに四塩化炭素を24週間以上反復投与すると、肝硬変を背景として肝細胞癌に類似した多発性肝腫瘍が発生する(図1)。また、マウス新生仔に変異原diethylnitrosamine (DEN)を投与すると数か月で多発性肝腫瘍が形成されるが、背景肝はほぼ正常である(図1)。我々はこれらの対称的な肝腫瘍モデルを用い、腫瘍の遺伝子発現プロファイルをcDNAマイクロアレイで調べた[9] 。その結果、H19、Igf2、Afp、Gpc3、Tff3を含む15個の腫瘍特異的遺伝子が同定された。さらに、四塩化炭素誘発腫瘍、DEN誘発腫瘍に加え、チオアセタミド(thioacetamide)反復投与による肝硬変を基盤とする肝腫瘍、背景肝に異常のない自然発生肝腫瘍を含め、これらの遺伝子の発現を定量RT-PCRで調べ、二次元階層クラスター解析を行った。その結果、正常対照肝、肝硬変、肝硬変に伴う肝腫瘍、正常肝に発生する肝腫瘍は異なったクラスターに分類されることが明らかになった(図2)。これは肝腫瘍の発生過程の違いにより、活性化される遺伝子が異なることを示している。興味深いことに、これらの遺伝子はほぼすべてが胎児期・新生児期の肝組織で強く発現しており[9] 、肝腫瘍の発生に伴い、脱分化が起こることが強く示唆された。
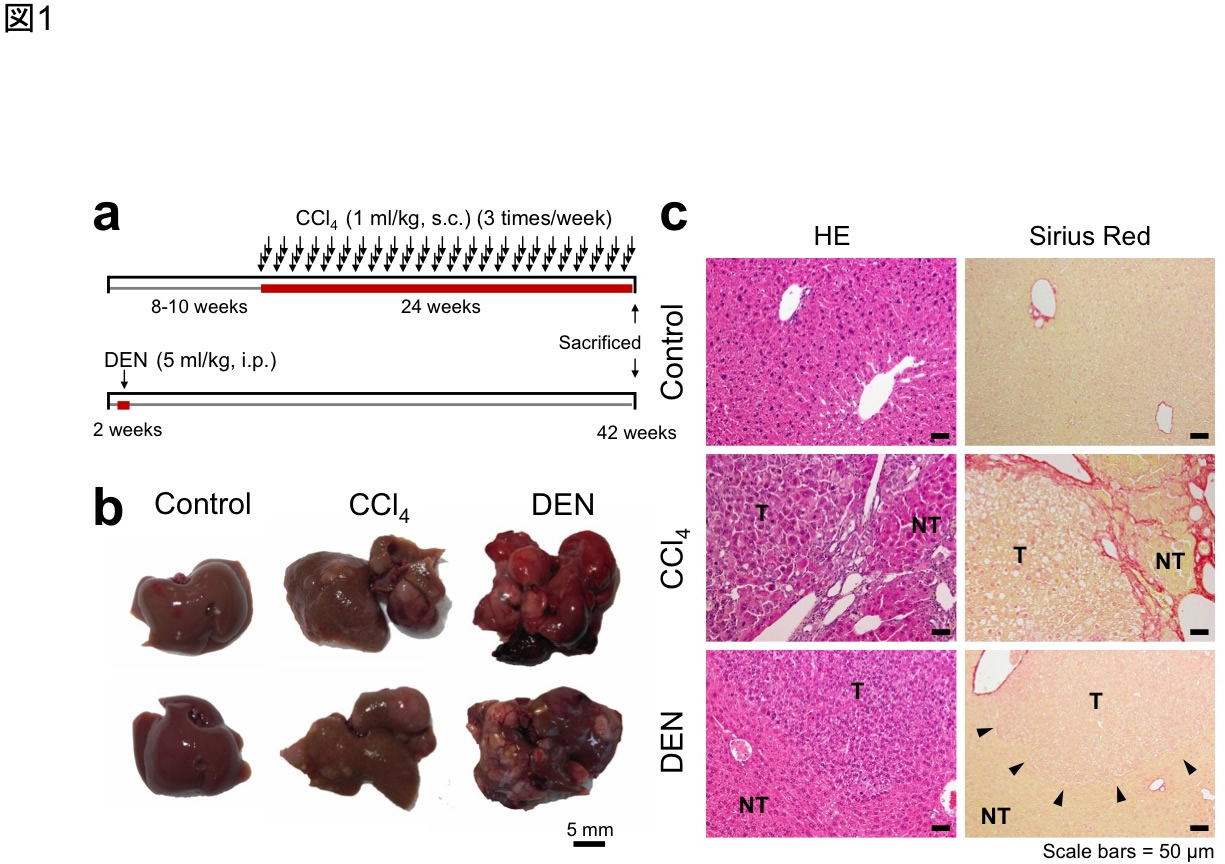
図1 慢性肝傷害または変異原により誘発されるマウス肝腫瘍
(a) 肝腫瘍誘発のプロトコール.雄性C57BL/6J×C3H F1マウスに対して,四塩化炭素を24週間にわたり反復投与する肝硬変モデルと生後2週間で変異原diethylnitrosamine (DEN)を1回投与する非肝硬変モデル.(b) 肝肉眼像.いずれのモデルでも多発性の肝腫瘍が認められる.Controlは正常肝.(c) 腫瘍の組織像(HE染色,シリウスレッド染色).いずれのモデルでも肝細胞性腫瘍(T)が形成されている.非腫瘍部(NT)は,四塩化炭素モデルではシリウスレッドで赤色に染色される結合織の豊富な肝硬変状態であるが,DENモデルではほぼ正常である.(Chen et al., Cancer Sci 2015;106:972-981より改編引用)
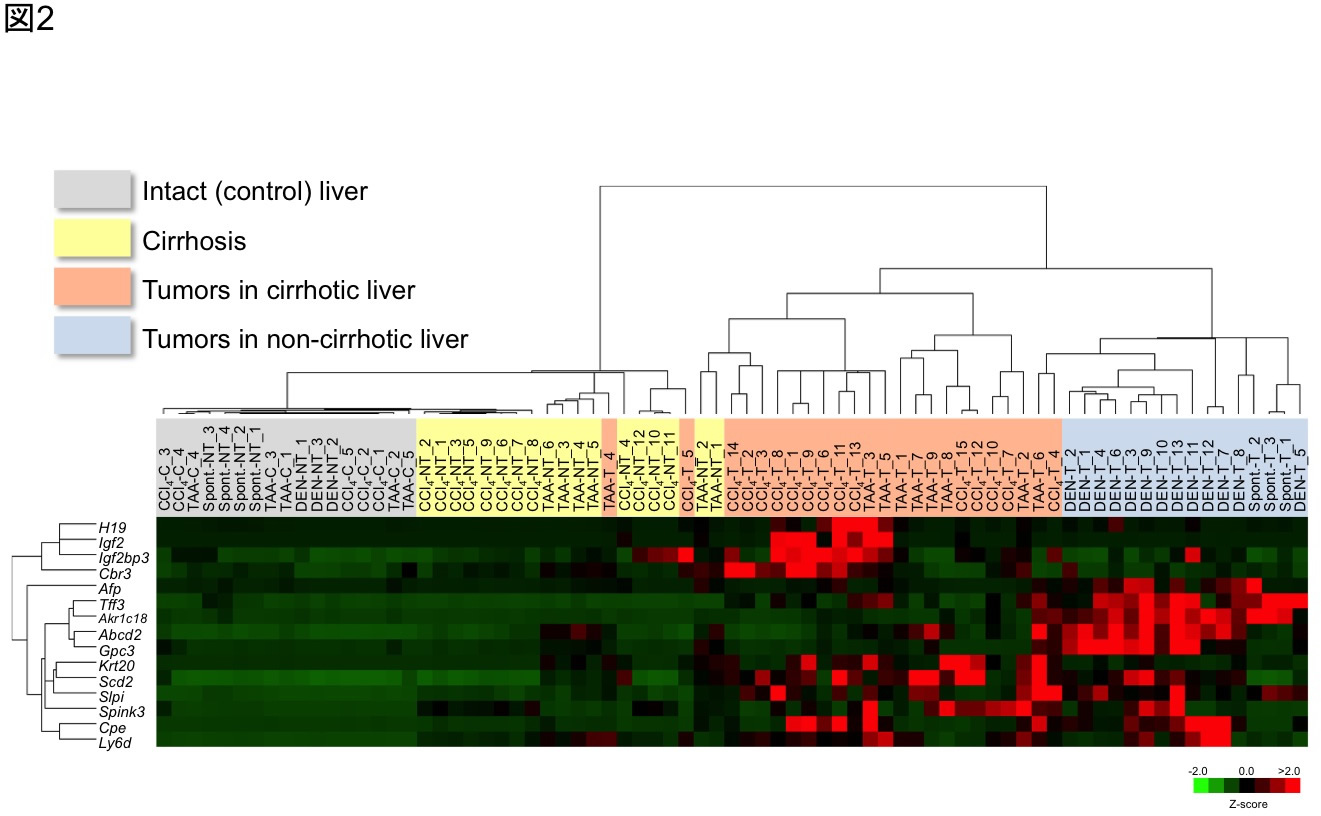
図2 マウス肝腫瘍における新生児期・胎児期遺伝子の発現
定量RT-PCRデータの二次元階層クラスター解析.正常肝,肝硬変(四塩化炭素反復投与),肝硬変モデルでの腫瘍(四塩化炭素またはチオアセタミド[thioacetamide, TAA]反復投与),非肝硬変モデル(DENまたは自然発生腫瘍)での15遺伝子の発現を示す.これらの遺伝子は四塩化炭素による肝硬変モデルの肝腫瘍,DENによる非肝硬変モデルの肝腫瘍のマイクロアレイ解析で同定した腫瘍特異的遺伝子で,新生児期・胎児期肝で強く発現している.(Chen et al., Cancer Sci 2015;106:972-981より改編引用)
肝腫瘍発生および脱分化におけるMyc遺伝子活性化の役割
Sleeping Beautyトランスポゾンシステムと尾静脈からのhydrodynamic injectionを組み合わせると、in vivoにおいて肝細胞にさまざまな癌遺伝子を導入し、短期間で肝腫瘍を誘導することが可能である[13]。この実験系では、大量(2.5 mL)のリンゲル液に溶解したプラスミドを急速に静注することにより、肝静脈と類洞内圧が上昇し、肝細胞に選択的に遺伝子が導入され、肝細胞ゲノム内の複数箇所に目的遺伝子が組み込まれる。ミリストイル型AKTと活性変異型HRASを同時に導入すると、約1か月で肝細胞癌が形成された(図3a)[10]。発癌過程の初期には細胞質に脂肪を蓄積した前癌細胞がみられるが、結節を形成する腫瘍細胞の多くは脂肪に乏しく、増殖性が高い(Ki67陽性核が多い)とともに、Mycの発現が認められた(図3b)。
肝細胞癌の発生におけるMycの活性化の意義を調べるため、Mycを競合的に阻害する癒合蛋白MadMyc(図4a)を発現するトランスポゾンベクターを活性型AKT/HRASととともに導入する実験を行った[10]。MadMycの同時発現は腫瘍の発生を顕著に抑制し(図4b)、この傾向はAKTに結合したhemagglutinin (HA)タグの免疫組織化学でも確認された(図4c, d)。
さらに、トランスポゾン法でMycを過剰発現させた場合の腫瘍表現型を検討した[10]。Mycのみを発現させても腫瘍は誘導されなかったが、Mycの導入はAKTやHRASによる腫瘍発生を劇的に促進した(図5a)。これらの腫瘍は肝細胞癌の組織学的特徴を示したが、Mycが導入された腫瘍では腫瘍細胞の核小体が大型化する傾向が認められた(図5b)。また、特にHRAS/Myc腫瘍では小型でN/C比が高い肝芽細胞様の腫瘍細胞が主体を占めており、脱分化が起こっていることが強く疑われた(図5a, b)[12]。実際、HRAS/Myc腫瘍では以前我々がマウス肝腫瘍モデルで同定したIgf2、H19、Afpなどの胎児期・新生児期遺伝子に加え、確立された肝芽細胞マーカーの1つであるDlk1の発現が特異的に認められた(図5c)。一方、AKTを同時に活性化するとこれらの遺伝子発現が抑制される傾向がみられた(図5c)。我々は最近、HRAS/Myc腫瘍ではDNA脱メチル化酵素Tet1のmRNA発現増加を伴うグローバル脱メチル化が起こると同時に、DNAメチル化酵素Dnmt1、Dnmt3のmRNA発現が増加することを見出した[12]。また、類似したエピゲノム調節酵素の発現変化やMyc高発現は初期の胎児肝においても認められ、脱分化型肝腫瘍は肝発生の初期過程を模倣していると考えられた[12]。

図3 トランスポゾンシステムによる肝細胞への癌遺伝子導入と腫瘍形成
(a) 活性型癌遺伝子(ミリストイル型AKTと活性変異型HRAS)の肝細胞への導入により誘発された肝腫瘍の肉眼像.雄性C57BL/6Jマウスの尾静脈から,Sleeping Beautyトランスポゼース発現ベクターを活性型癌遺伝子を挿入したトランスポゾンカセットベクターとともに急速に注射すると,肝細胞ゲノムに癌遺伝子が組み込まれる.(b) AKT/HRAS腫瘍の組織像(HE染色)およびMyc,Ki-67の免疫組織化学.2週間後では脂肪を蓄積した前癌細胞が小葉内に多数観察される(白矢頭).その後,脂肪蓄積を伴わない細胞(黒矢頭)による腫瘍結節(T)が形成される.これらの細胞の核にはMyc発現がみられ,Ki-67陽性率が高い.(Xin et al., Oncogene 2017;36:5087-5097より改編引用)

図4 Myc阻害蛋白発現によるAKT/HRAS腫瘍の抑制
(a) MadMycの構造の模式図.MadMycは競合阻害によりMycの働きを抑制する.(b) MadMycによるAKT/HRAS腫瘍の抑制.遺伝子導入8週間後の肝肉眼像.MadMycを挿入したトランスポゾンカセットベクターをAKT,HRASと同時に注射した場合,腫瘍はほとんど形成されない.(c) 肝組織像(HE染色)およびhemagglutinin (HA)免疫組織化学(導入したAKTのHAタグを検出する).対照群ではHA陽性の腫瘍細胞の増殖がみられるが,MadMycを導入するとHA陽性細胞が小葉内に散見されるのみである.(d) HA陽性領域の定量結果.(Xin et al., Oncogene 2017;36:5087-5097より改編引用)
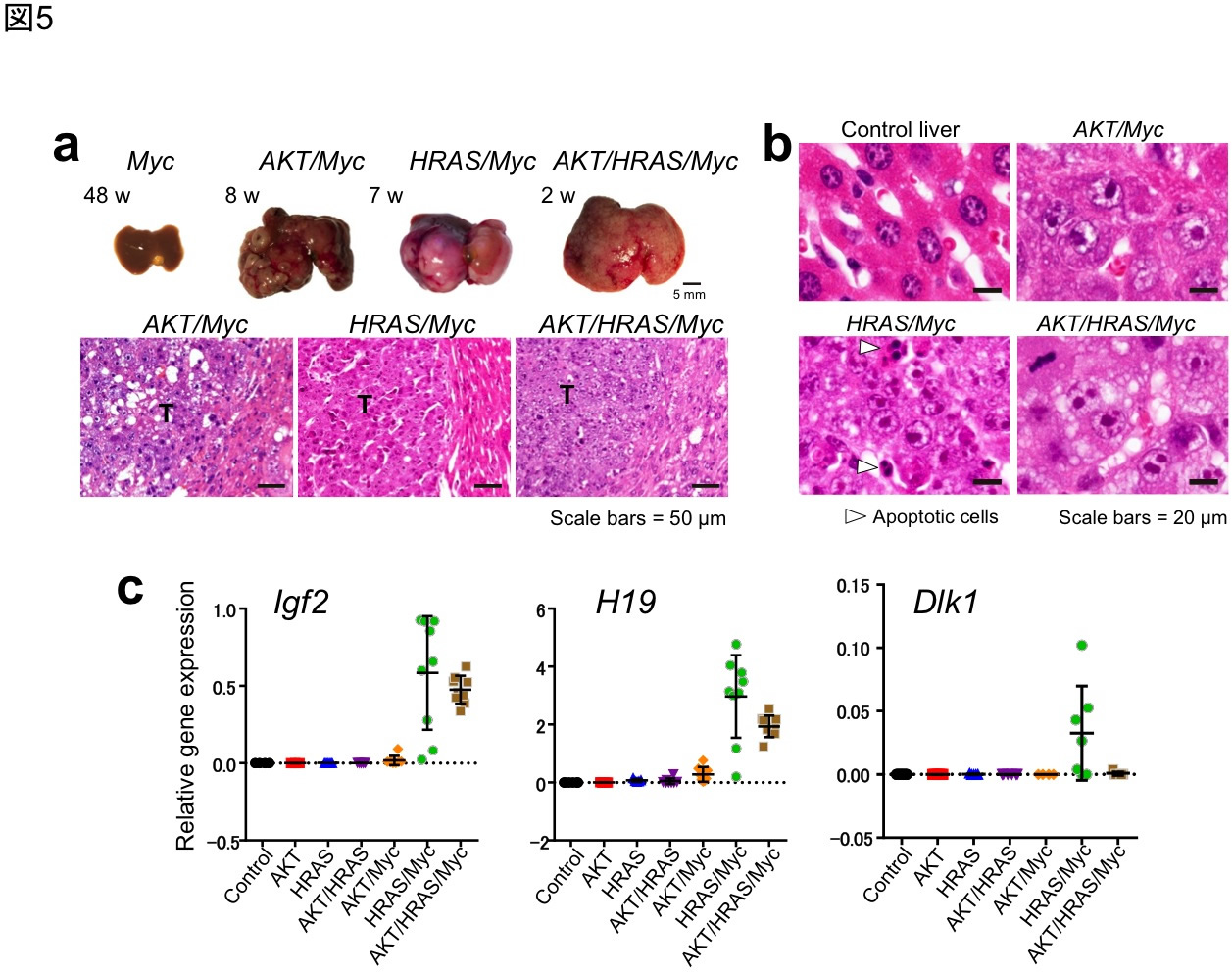
図5 Myc過剰発現による肝腫瘍発生の促進と脱分化形質の誘導
(a) Myc過剰発現によるAKT,HRAS,AKT/HRAS腫瘍の発生促進.肝肉眼像および組織像(HE染色).Myc単独では腫瘍形成はみられない.腫瘍(T)は肝細胞癌の像を示すが,HRAS/Myc腫瘍は特に細胞密度が高い.(b) 肝腫瘍組織像(HE染色,高倍率).Mycを導入した腫瘍ではいずれも核内に腫大した核小体が認められる.HRAS/Myc腫瘍では腫瘍細胞のアポトーシスが高頻度にみられる(白矢頭).(c) 種々の癌遺伝子により誘導された肝腫瘍におけるIgf2,H19,Dlk1のmRNA発現の定量RT-PCR解析. HRAS/Myc腫瘍ではこれらの遺伝子が特異的に発現しており,脱分化が起こっていることが示唆される.AKT/HRAS/Myc腫瘍でもIgf2やH19の発現はみられるが,Dlk1は発現しない.(Xin et al., Oncogene 2017;36:5087-5097およびWatanabe et al., Hepatology Communications 2019, in pressより改編引用)
肝細胞性腫瘍の表現型決定における癌遺伝子シグナリングの相互作用
次に我々は、腫瘍発生に深く関わることが知られているHippo-YAP経路が肝細胞性腫瘍の表現型決定にどのような役割を担っているかを検討した[11] 。活性型AKTとYAPの導入では間質増生とcytokeratin 19 (CK19)発現を伴う胆管癌が誘導された(図6a)。肝細胞特異的にCreを発現させることが可能なAAV8-Tbg-Creを感染させたCreレポーターマウス(ROSA26R)で同様に腫瘍を誘導し、腫瘍の胆管構造が既存の胆管上皮細胞ではなく、肝細胞に由来することが確認された(図6b)。また、YAPとMycの組み合わせでは興味深いことに、肝細胞癌と胆管癌の混在した混合型肝癌に類似した腫瘍が誘導されることが明らかになった(図6a)。YAP/Myc腫瘍ではDlk1やAFPの蛋白発現やmRNA発現も認められたが、HRAS/Myc腫瘍の場合と同様にAKTの同時活性化はこれらの発現を抑制した(図6a, c)。
Notch経路は、肝発生過程における胆管上皮細胞の分化に重要な役割を担っている[14]。Notch intracellular domain (NICD)導入によるNotch経路の活性化を活性型AKTとMycの導入とともに行うと、AKT/YAPの場合に較べ、より異型度が高い胆管癌が誘導された[11] 。さらに、NICDと活性型HRASにより誘導された腫瘍は、間葉系細胞のマーカーであるvimentinを発現する紡錘形細胞のびまん性増殖からなる非上皮性悪性腫瘍の組織像を示し、癌遺伝子活性化による上皮-間葉転換により肝細胞から肉腫様肝癌が発生することが示唆された(Yamamoto et al., manuscript in preparation)。
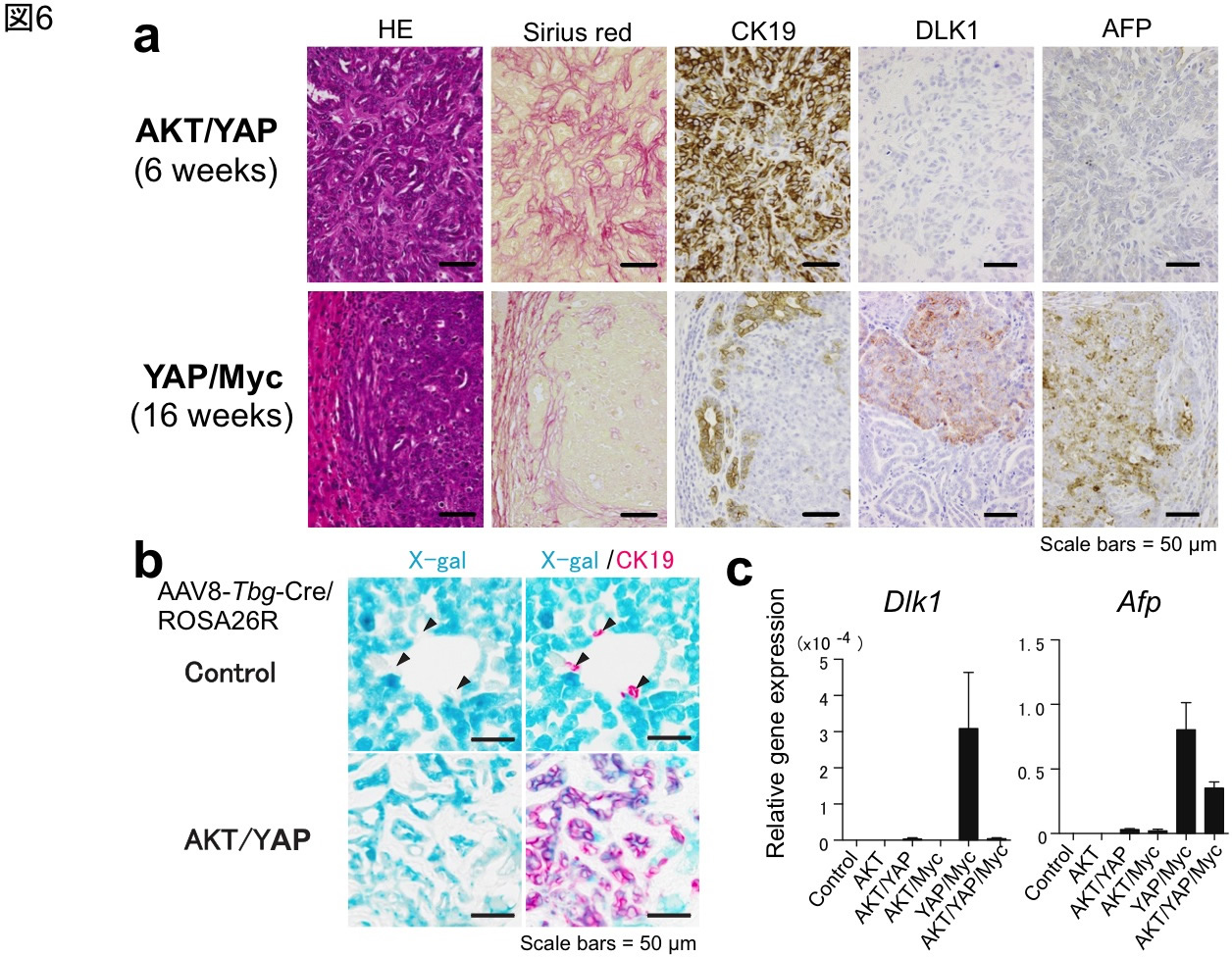
図6 活性型YAP発現による肝細胞からの胆管癌,混合型肝癌の発生
(a) AKT/YAP,YAP/Mycの組み合わせにより誘導された肝腫瘍の組織像(HE染色,シリウスレッド染色)およびcytokeratin 19 (CK19),DLK1,AFP免疫組織化学.AKT/YAP腫瘍は不規則な管腔構造を示し,シリウスレッド陽性の強い間質増生を伴っている.また,胆管上皮細胞のマーカーCK19が陽性である.YAP/Myc腫瘍ではCK19陽性の胆管癌と肝細胞癌が混在しており,一部にDLK1やAFPの陽性像が認められる. (b) AAV8-Tbg-Creを感染させたROSA26RマウスにおけるAKT/YAP腫瘍のX-gal染色およびX-gal/CK19二重染色.胆管癌がX-gal陽性の肝細胞に由来していることがわかる.矢頭は既存の胆管構造を示す(X-gal陰性、CK19陽性)。(c) AKT,YAP,Mycの組み合わせにより誘導された肝腫瘍におけるDlk1,Afp mRNA発現の定量RT-PCR解析.YAP/Myc腫瘍では両者の遺伝子発現が認められる.(Yamamoto et al., Am J Pathol 2017;187:2711-2725より改編引用)
おわりに
これまで我々は肝上皮系細胞の相互可塑性、特に成熟肝細胞の胆管上皮細胞への分化転換を中心に研究を進めてきた。肝細胞表現型の可塑性は、肝細胞性腫瘍の表現型の多様性を理解する上で重要であると考えられる。我々はまだ肝癌発生に関わる癌遺伝子シグナル経路のごく一部のみを解析したにすぎないが、これらの経路の相互作用により肝細胞から肝細胞癌だけでなく胆管癌や混合型肝癌、さらに肝芽腫様の脱分化型肝癌が誘導されることが明らかになった(図7)。胆管癌が胆管上皮細胞から発生しうることは実験的に示されているが[15,16] 、現時点では、肝内胆管癌には胆管上皮細胞、肝細胞由来のものが存在しうると想定しておく必要がある。また、肝芽細胞方向への脱分化にはMycの活性化が重要であること、PI3K-AKT経路は腫瘍の悪性度を高める一方、脱分化を抑制する傾向があることが明らかになった。これは、腫瘍の悪性度の指標としての「分化度の低さ(低分化)」を「脱分化」と区別する必要があることを示唆している。最近、肝微小環境が肝腫瘍の表現型を規定する可能性が報告されており[17] 、今後、腫瘍細胞内のシグナル経路の変化が微小環境にどのような影響を及ぼすかについても検討を進めていく必要がある。
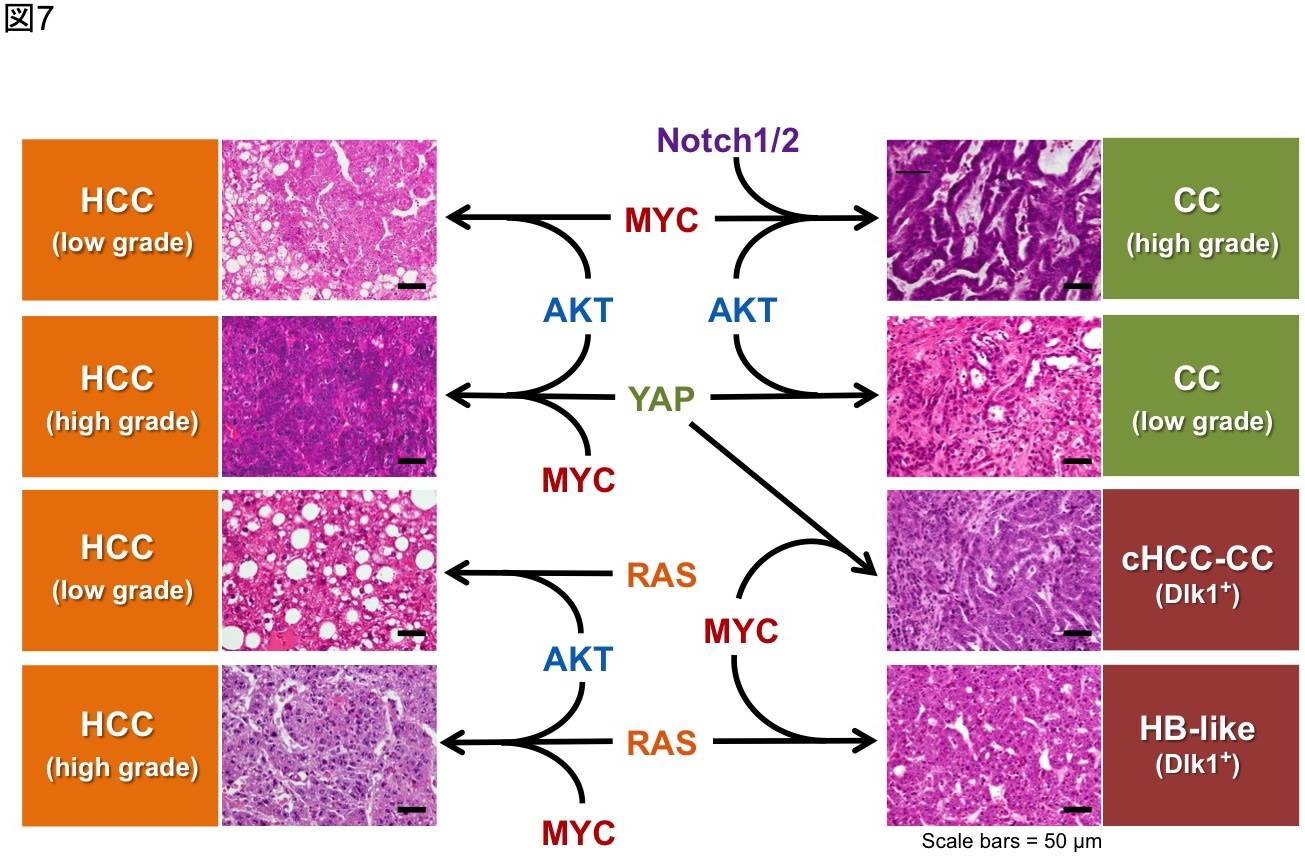
図7 癌遺伝子活性化とマウス肝腫瘍の表現型の関連性
トランスポゾンシステムとhydrodynamic injectionを用いたマウス肝発癌モデルを用い,活性化する癌遺伝子の種類と組み合わせにより,肝細胞から肝細胞癌(HCC)だけでなく,胆管癌(CC),混合型肝癌(cHCC-CC),肝芽腫様腫瘍(HB-like)が発生しうる.肝細胞性腫瘍の表現型の多様性は,分化転換や脱分化を含む肝細胞形質の可塑性をもとに理解することが可能である.
謝辞
本稿で紹介させていただいた研究の多くは、私の前任地の秋田大学大学院医学系研究科分子病態学・腫瘍病態学講座で得られた知見を基盤として、旭川医科大学病理学講座腫瘍病理分野のこれまでのスタッフとともに進めてきたものです。研究に貢献してくれたすべての皆様にこの場をお借りして御礼を申し上げます。
文献
- Brunt E, Aishima S, Clavien PA, Fowler K, Goodman Z, Gores G, Gouw A, et al. cHCC-CCA: Consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology 2018;68:113-126.
- Shiojiri N. The origin of intrahepatic bile duct cells in the mouse. J Embryol Exp Morphol 1984;79:25-39.
- Nishikawa Y, Tokusashi Y, Kadohama T, Nishimori H, Ogawa K. Hepatocytic cells form bile duct-like structures within a three-dimensional collagen gel matrix. Exp Cell Res 1996;223:357-371.
- Nishikawa Y, Doi Y, Watanabe H, Tokairin T, Omori Y, Su M, Yoshioka T, et al. Transdifferentiation of mature rat hepatocytes into bile duct-like cells in vitro. Am J Pathol 2005;166:1077-1088.
- Nagahama Y, Sone M, Chen X, Okada Y, Yamamoto M, Xin B, Matsuo Y, et al. Contributions of hepatocytes and bile ductular cells in ductular reactions and remodeling of the biliary system after chronic liver injury. Am J Pathol 2014;184:3001-3012.
- Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, Umemura A, Taniguchi K, et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 2015;162:766-779.
- Tanimizu N, Ichinohe N, Yamamoto M, Akiyama H, Nishikawa Y, Mitaka T. Progressive induction of hepatocyte progenitor cells in chronically injured liver. Sci Rep 2017;7:39990.
- Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911-2915.
- Chen X, Yamamoto M, Fujii K, Nagahama Y, Ooshio T, Xin B, Okada Y, et al. Differential reactivation of fetal/neonatal genes in mouse liver tumors induced in cirrhotic and non-cirrhotic conditions. Cancer Sci 2015;106:972-981.
- Xin B, Yamamoto M, Fujii K, Ooshio T, Chen X, Okada Y, Watanabe K, et al. Critical role of Myc activation in mouse hepatocarcinogenesis induced by the activation of AKT and RAS pathways. Oncogene 2017;36:5087-5097.
- Yamamoto M, Xin B, Watanabe K, Ooshio T, Fujii K, Chen X, Okada Y, et al. Oncogenic Determination of a broad spectrum of phenotypes of hepatocyte-derived mouse liver tumors. Am J Pathol 2017;187:2711-2725.
- Watanabe K, Yamamoto M, Xin B, Ooshio T, Goto M, Fujii K, Liu Y, Okada Y, Furukawa H, Nishikawa Y. Emergence of the dedifferentiated phenotype in hepatocyte-derived tumors in mice: Roles of oncogene-induced epigenetic alterations. Hepatology Communications 2019, in press (https://doi.org/10.1002/hep4.1327).
- Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014;184:912-923.
- McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002;129:1075-1082.
- Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, Bessho K, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest 2014;124:3241-3251.
- Ikenoue T, Terakado Y, Nakagawa H, Hikiba Y, Fujii T, Matsubara D, Noguchi R, et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci Rep 2016;6:23899.
- Seehawer M, Heinzmann F, D'Artista L, Harbig J, Roux PF, Hoenicke L, Dang H, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018;562:69-75.

