





研究交流
異なる病因の慢性肝障害における成体肝前駆細胞の制御機構
田中 稔
国立国際医療研究センター研究所
はじめに
げっ歯類を用いた重篤または慢性的な肝障害モデルにおいて、門脈周囲に出現するオーバル細胞と呼ばれる小型で増殖性の高い細胞は、肝再生に関わる肝前駆細胞(Liver Progenitor Cell、以下、LPCと呼ぶ)であるとされてきた(1)。LPCは既存の胆管と多くのマーカー分子を共有し、LPCが増殖する様子は、まるで胆管が延びてくるように見えることから、偽胆管増生や細胆管反応などとも呼ばれている。LPCは肝実質細胞(肝細胞)と胆管上皮細胞の二方向性に分化しうる細胞と定義されるが、近年の細胞系譜解析(Lineage Tracing)を用いた多くの報告から、LPCの肝再生への寄与は限定的であるとする意見が大勢を占めるようになった(2,3)。これに対して、最近、S. Forbesらのグループは、肝細胞に増殖抑制等の負荷がかかった状態では、LPCは十分に肝再生に寄与しうるという新たな見解を示している(4)。これらの事実から見えてきたものは、背景となる肝障害の“質”がその後のLPCによる再生様式に大きな影響を及ぼしうるということである。しかし、従来のLPC研究では、使用する肝障害モデルの病因についてはあまり考慮されておらず、異なる病態下におけるLPCの制御機構についてもほとんど明らかとなっていない。我々は最近、肝障害モデルに応じてLPC上で発現が変動する新規マーカー分子を同定し、その発現の違いが障害後のLPCの性状を規定していることを明らかにした(5)。本稿ではその内容についてデータを交えて紹介したい。なお、肝再生に関わるLPCについては近年様々な細胞ソースが報告されていることから、本稿で取り上げるLPCは細胆管反応に関わる胆管様の細胞とさせていただく。
LPCの性状は肝障害モデルにより異なる
マウスを用いたLPC研究では、コリン欠乏エチオニン添加(CDE)食またはDDC(3,5-diethoxycarbonyl-1,4-dihydrocollidine)含有食を長期投与する慢性肝障害モデルが主に用いられてきた(6,7)。しかし、両モデルでは病因が全く異なっており、CDEモデルは脂肪性肝炎様の病態を呈し、主に肝細胞が障害されるのに対して、DDCモデルは原発性硬化性胆管炎モデルとされ、主に胆道が障害され、著しい胆汁うっ滞を伴う(8,9)。いずれのモデルでもLPCは効率よく誘導されるが、増殖してくるLPCの性状は必ずしも一様ではなく、LPCマーカーの一つであるEpCAMで免疫染色すると、明らかな形態の違いが見てとれる(図1A)。CDEモデルのLPCは主に紡錘形の形態を呈し、門脈付近から実質域へと浸潤して高い運動性を示すのに対し、DDCモデルのLPCは明瞭な管腔構造を呈し、門脈周囲で無数の小葉間胆管様の形態をとっていることがわかる。障害間でこのような違いがあるにも関わらず、LPCの運動性や管腔形成能を規定する分子メカニズムはこれまで一切不明であった。なぜなら、従来の研究ではLPCは胆管マーカーで一様に染色される集団として捉えられるのみであり、病因の違いに応じてLPCで異なる発現様式を示すような分子の存在が知られていなかったからである。
我々は以前に、マウス胎仔期の肝芽細胞にLutheran(Lu)という分子が発現することを見出していた。Luは別名Basal Cell Adhesion MoleculeまたはCD239と呼ばれるI型の膜タンパク質であり、ラミニンα5(Lama5)鎖に結合する分子として知られていた(10)。今回、障害肝におけるLuの発現を免疫組織化学染色およびFACS解析で調べたところ、CDEモデルのLPCではLuが高発現しているのに対し、DDCモデルでは逆に発現が低下していることを見出した(図1B, C)。そこで、CDEモデルの障害肝よりLu強陽性とLu陰性のLPCをそれぞれ単離し、運動性や管腔形成能におけるLu発現の意義について検討を行なうことにした。
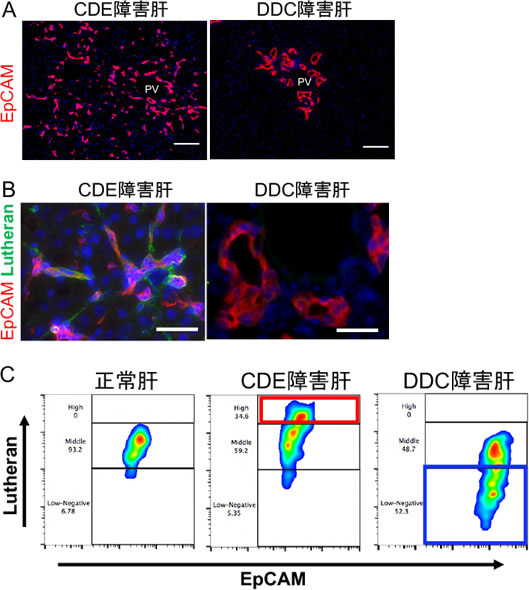
図1. CDEおよびDDCモデルの障害肝におけるLPCの形態とLuの発現
(A) 各障害肝(3週間投与後)の抗EpCAM抗体による免疫染色像.(B) 抗EpCAM抗体と抗Lutheran抗体による共染色像.(C) 抗EpCAM抗体と抗Lutheran抗体による非実質細胞のフローサイトメトリー解析.図はEpCAMゲート後のプロットを示す.PV:門脈. Scale Bar: 100μm.(文献5より一部改変)
Luの発現がLPCの性状を規定する
まず、単離したLPCをディッシュ上に播種したところ、Lu陰性LPCは典型的な上皮細胞様の形態を示したのに対し、Lu陽性LPCでは葉状仮足が認められた(図2A)。次に、スクラッチアッセイにより運動能を調べた結果、Lu陽性LPCの方がLu陰性LPCよりも高い運動性を示した(図2B)。一方、管腔形成能については、コラーゲン-マトリゲルを用いた三次元培養によるシスト(嚢胞)形成という形で評価した結果(11)、Lu陰性LPCは高いシスト形成能を示したのに対し、Lu陽性LPCは凝集塊を作るのみであった(図2C)。よって、Lu陽性及び陰性のLPCは、障害肝から単離後も肝臓内での形質を維持していることが示された。そこで、Luの発現自体が運動性や管腔形成能を規定しているのかについて、さらに検討を行なった。Lu陰性のLPCにレトロウイルスベクターを用いてLuを強制発現させたところ、運動性を獲得するとともに管腔形成能を失った(図2D)。すなわち、Luの発現がLPCの性状を規定していることが明らかとなった。
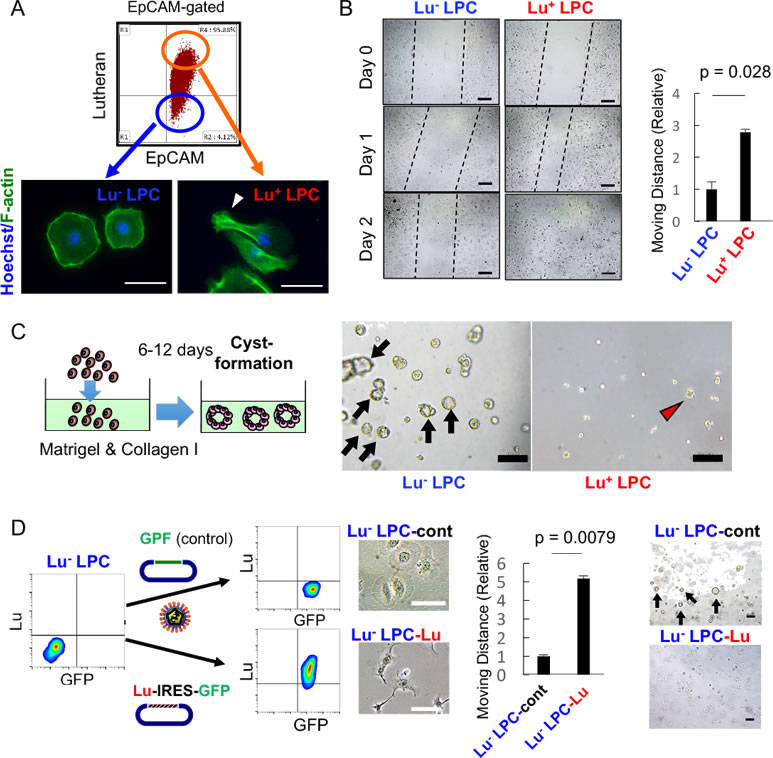
図2. Lu発現によるLPCの運動性および管腔形成能の制御
(A) CDE障害肝のFACS解析によるLu陽性およびLu陰性LPCの単離. 図は非実質細胞のEpCAMゲート後のプロットを示す.矢尻: 葉状仮足.(B) スクラッチアッセイによるLPCの運動性の評価.(n=4) (C) 三次元培養系によるシスト形成アッセイと管腔系性能の評価.(D) レトロウイルスベクターを利用したLu陰性LPCへのLuの強制発現とその後の性状解析.ソーティング後の細胞の培養中の形態(左図)とスクラッチアッセイ (n=5) 及びシスト形成アッセイによる比較(右図).Scale Bar: 100μm.(文献5より一部改変)
LPCにおけるLuの作用機序
LPCの性状がLuの発現によって規定されていたことから、次にその作用機序について検討を行なった。前述したようにLuはLama5鎖に結合できるため、Lama5を含むラミニン511の受容体として機能する。一方、ラミニン511はインテグリンα3β1またはα6β1のリガンドとして機能することから(12)、我々は、「Luがインテグリンに対して競合的に働き、ラミニン511からのインテグリンシグナルを減弱化させることでLPCの性状を規定している。」という仮説を立てた(図3A)。もし、この仮説が正しいとするならば、インテグリンβ1に対する中和抗体を作用させることで、Lu陰性のLPCのフェノタイプをLu陽性タイプに転換できると考えた。実際に行なってみると、Lu陰性LPCは運動性を獲得し、シスト形成能を失ったことから、Luはインテグリンシグナルを調節することで障害後のLPCの性状を規定していることが強く示唆された(図3B, C)。なお、Lama5はCDE障害肝、DDC障害肝のいずれにおいてもLPC自体が発現していることも明らかにしている。
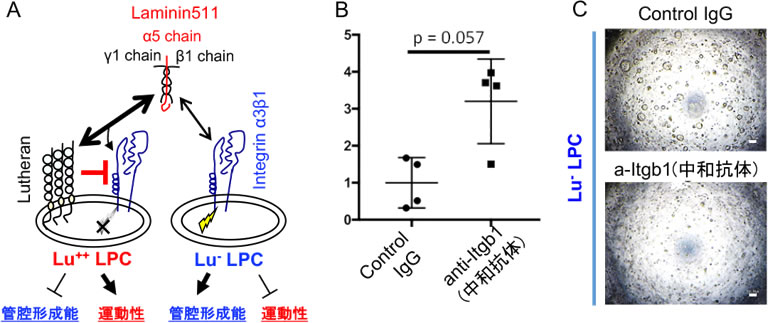
図3. LuのLPCに対する作用機序の検討
(A) ラミニン/インテグリン シグナルに対するLu作用の作業仮説.(B, C) 抗インテグリンβ1中和抗体添加によるLu陰性LPCの性状の変化.スクラッチアッセイ(B)とシスト形成アッセイ(C)の結果を示す.抗体の添加によりLPCは運動性を獲得し、シスト形成能を喪失した.Scale Bar: 100μm.(文献5より一部改変)
Lu ノックアウトマウスでは細胆管反応が著しく障害される
In vitroの実験結果から、LuがLPCの性状を規定する可能性が示されたが、実際にLuがin vivoにおいてもLPCの制御に関わるのか否かについて調べるために、CRISPR/Cas9法を用いてノックアウト(KO)マウスの作製を行なった(図4A)。CDEモデルで比較した結果、野生型マウスではLPCが実質域深く浸潤するのに対して、KOマウスでは門脈周囲に留まる様子が観察された(図4B)。この際、増殖マーカーであるKi67の陽性率に有意差は認められておらず、Lu欠損は増殖性ではなく、運動性に対する障害であることを確認している。以上の結果から、Luは異なる肝障害に応じてLPCの性状を規定し、肝リモデリングを制御する必須の因子であることが明らかとなった(図4C)。
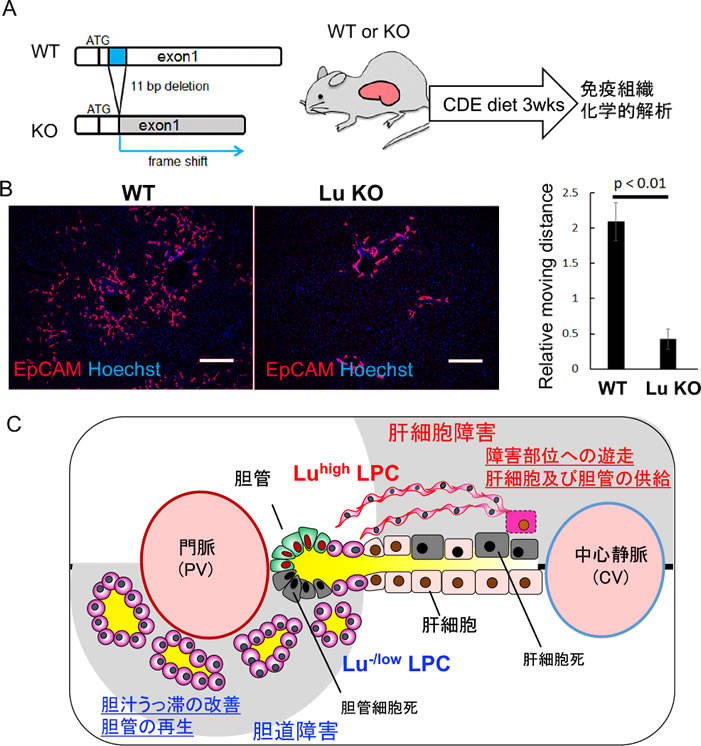
図4. KOマウスを用いたin vivoでのLuの機能解析
(A) CRISPR/Cas9法によるKOマウスの作製とCDEモデルによる評価.(B) 野生型(WT)またはKOマウスのCDE障害肝(3週間投与後)の抗EpCAM抗体による免疫染色像(左図)とLPCの門脈からの移動距離の比較(右図). (n=5) (C) 異なる肝障害におけるLuによる肝リモデリング制御のモデル図.(文献5より一部改変)
おわりに
マウスのLPC研究でCDEやDDCモデルが主に用いられてきたのは、単に細胆管反応が起こりやすいからという理由に他ならず、その病因や障害の背景にまで言及している論文は数少ない。CDEモデルとDDCモデルでは主に障害を受ける細胞種が異なるため、出現するLPCの性状も分化の方向性も違って当然と考えられる。本研究では、これまで異なる肝障害モデルで一様に扱われてきたLPCに明確な違いがあることを分子レベルで明らかにした。一方、LPCの概念の元となるラットのオーバル細胞は、薬剤で肝細胞の増殖を抑制した上で、肝切除や肝細胞障害を与えることで出現、増殖する細胞であった(13,14)。すなわち、肝細胞の増殖による通常の再生ができないため、ラットオーバル細胞には肝再生に寄与する合目的性があった。しかし、マウスのCDEモデルもDDCモデルも残存する肝細胞は増殖することができる状況にある。このような病態下では細胞系譜解析においてLPCの潜在能力を十分に評価できていたとは考えにくい。実際、前述したように、肝細胞の増殖を抑制した状態ではマウスLPCも十分に肝再生に寄与できることが報告されており(4)、マウスLPC研究は、紆余曲折はあったものの長い時間を経て、ラットで見出されたオーバル細胞の原点に戻りつつある。それゆえ、マウスLPCによる肝再生機構の研究は新たなステージに入ったと言え、今後は肝障害の背景や重篤度、細胞死など様々な要素を考慮した上でLPCのキャラクタライズが進んで行くものと思われる。本研究がその一助となれば幸いである。
謝辞
最後に、本研究は早稲田大学 先端理工学部の三浦泰史君が当研究室に在籍中に主に行なった研究成果ですが、兵庫医科大学の大村谷昌樹先生、金沢大学の原田憲一先生、東京薬科大学の吉川大和先生、東京大学の宮島篤先生など多くの先生方のご協力のもと行ないました。この場を借りて御礼申し上げます。
参考文献
- Miyajima A, Tanaka M, Itoh T. 2014. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 14:561–574.
- Malato Y, Naqvi S, Schürmann N. et al. 2011. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Invest. 121:4850–4860.
- Yanger K, Knigin D, Zong Y. et al. 2014. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell 15:340-349.
- Raven A, Lu WY, Man TY. et al. 2017. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 547:350–354.
- Miura Y, Matsui S, Miyata N. et al. 2018. Differential expression of Lutheran/BCAM regulates biliary tissue remodeling in ductular reaction during liver regeneration. eLife 7:e36572.
- Akhurst B, Croager EJ, Farley-Roche CA. et al. 2001. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology 34: 519–522.
- Preisegger KH, Factor VM, Fuchsbichler A. et al. 1999. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab. Invest. 79:103–109.
- Aharoni-Simon M, Hann-Obercyger M, Pen S. et al. 2011. Fatty liver is associated with impaired activity of PPARg-coactivator 1a (PGC1a) and mitochondrial biogenesis in mice. Lab. Invest. 91: 1018–1028.
- Fickert P, Stöger U, Fuchsbichler A. et al. 2007. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am. J. Pathol. 171:525–536.
- Parsons SF, Lee G, Spring FA. et al. 2001. Lutheran blood group glycoprotein and its newly characterized mouse homologue specifically bind alpha5 chain-containing human laminin with high affinity. Blood 97:312–320.
- Tanimizu N, Miyajima A, Mostov KE. 2007. Liver progenitor cells develop cholangiocyte-type epithelial polarity in three-dimensional culture. Mol. Biol. Cell. 18:1472–1479.
- Kikkawa Y, Sasaki T, Nguyen MT. et al. 2007. The LG1-3 tandem of laminin alpha5 harbors the binding sites of Lutheran/basal cell adhesion molecule and alpha3beta1/alpha6beta1 integrins. J. Biol. Chem. 282:14853–14860.
- Evarts RP, Nagy P, Nakatsukasa H. et al. 1989. In vivo differentiation of rat liver oval cells into hepatocytes. Cancer Res. 49:1541–1547.
- Fausto N, Campbell JS. 2003. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech, Dev. 120:117-130.

