





ホットトピックス
低酸素応答による「乳酸からの糖新生」の制御
〜その分子メカニズムの解明から乳酸アシドーシス治療法の開発へ〜
南嶋洋司
九州大学 生体防御医学研究所 細胞機能制御学部門
分子医科学分野 特任准教授
【はじめに】
利用できる酸素が減少した「低酸素環境」に対する我々の身体の応答反応(低酸素応答)は、主に低酸素応答のマスターレギュレーターとも呼ばれる転写因子HIF (hypoxia-inducible factor)によって制御されているのだが、そのHIFもまたプロリン水酸化酵素PHD (prolyl hydroxylase domain-containing protein)によって負に制御されているため[1] 、PHDは酸素濃度センサーとして低酸素応答を制御していると言うことができる (Figure 1) 。本稿では、PHD (PHD1~3)を介した低酸素応答によるエネルギー代謝制御機構のなかでも、特に乳酸代謝に関する筆者らの研究をご紹介したい。
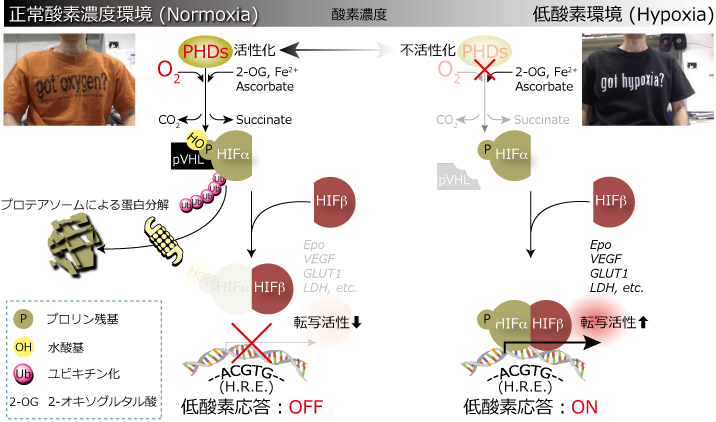
Figure 1. PHD-HIFを介した低酸素応答の分子メカニズム
PHDの酵素活性が低酸素応答をONにするかOFFにするかを決定している。
【低酸素応答による解糖の活性化】
PHDは酵素学的には二価の鉄イオンと2-オキソグルタル酸(α-ケトグルタル酸)依存的ジオキシゲナーゼ(酸素添加酵素の一種)であるが、酸素添加酵素は(酸素分子に対するKm値にも依るが)酸素濃度が低下するとその酵素活性が低下する。低酸素などによって細胞内でPHDの活性が低下すると、HIFのα-サブユニット (HIFα)内の特定のプロリン残基の水酸化を指標としたHIFαのユビキチン-プロテアソーム依存的蛋白分解が生じなくなるため、HIFαが細胞内に急速に蓄積する。こうして活性化した転写因子HIFは解糖に関与するトランスポーターや酵素の発現を誘導するため、細胞に取り込まれたグルコースはピルビン酸まで代謝されたあと、アセチルCoAではなく乳酸へと変換される。即ちPHDの活性をOFFにすると、大量の乳酸が細胞外に放出されることになる (Figure 2A)[2]。
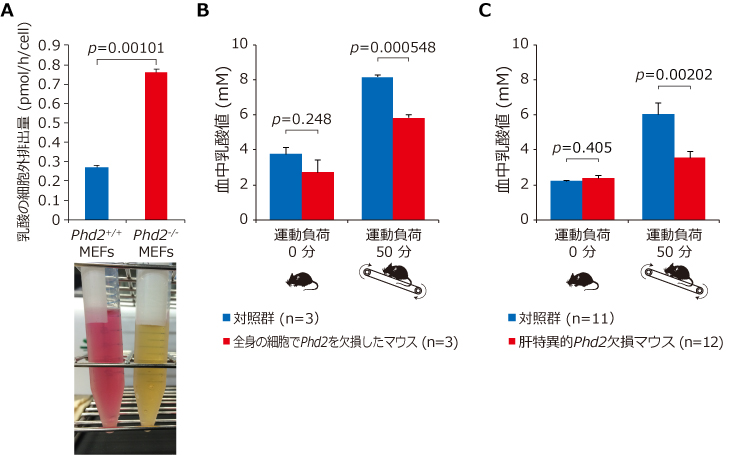 Figure 2. 低酸素応答と乳酸代謝
Figure 2. 低酸素応答と乳酸代謝
(A) Phd2を欠損したマウス胎仔線維芽細胞 (MEFs)では解糖が活性化し、大量の乳酸が細胞外に放出される。(B) タモキシフェン投与により全身の細胞でPhd2を破壊したマウスにおいては、予想に反して対照群よりも血中乳酸値が低い。(C) 肝特異的Phd2欠損マウスにおいても同様。すなわち、肝臓でPHD2を阻害すれば運動などの生理的乳酸負荷時の血中乳酸値を低下させることができることがわかる(文献5より改変)。
【肝における低酸素応答による乳酸からの糖新生の活性化】
では、もしここで、仮に全身の細胞でPHDを介した低酸素応答がONになったらどうなるであろうか?全身の細胞から大量の乳酸が血中に放出されるのである、個体はきっと重度の乳酸アシドーシス[3]に陥ってしまうであろう。
そこで、CAGG-CreERTMシステムを用いてタモキシフェン投与によって全身の細胞でPhd2(3つある哺乳動物のPHD遺伝子のなかで主要なプロリン水酸化酵素)を破壊することができるマウスにおいて血中乳酸値を測定してみた。すると、驚くべきことに、全身の細胞でPhd2が破壊されHIFが活性化し解糖が亢進して血中に大量の乳酸が放出されているはずのPhd2ノックアウトマウスにおいては、血中乳酸値が対照群と比較して逆に低いことがわかった(特に運動負荷後のように血中乳酸値が上昇する状況においてその傾向が顕著であった) (Figure 2B)。Phd2ノックアウトマウスにおける尿中乳酸値は対照群と同レベルであったため、“腎臓においてPhd2遺伝子が機能しなくなることで尿中への乳酸排出が増加した結果血中乳酸値が低下した”訳では無いこともわかった。これは、in celluloの結果から予想されたin vivoの結果が予想と真逆になるという、非常に興味深い現象であった。
1928年にCori夫妻によって「解糖によって骨格筋で産生された乳酸は血流に乗って肝臓へ運ばれ糖新生の原料として再利用され、肝で新生されたグルコースは再び血流に乗って筋肉へ運ばれ、解糖に利用される」という、炭素のリサイクル機構“Cori回路”が提唱された[4]。即ち、血中乳酸値はCori回路によって制御されているとも言える。そこで筆者らは、「低酸素応答は末梢組織においては解糖を活性化するが、肝臓では解糖ではなく糖新生を活性化させるのではないか?」と考えた。その仮説を立証するため、Albumin-Creシステムを用いて肝特異的にPhd2遺伝子を破壊したマウスを作製して血中乳酸値を測定してみたところ、予想通り肝特異的Phd2欠損マウスの血中乳酸値は対照群よりも低値であった (Figure 2C)。
この肝特異的Phd2欠損マウスにおいては、対照群と比較して乳酸からの糖新生に必要なトランスポーターや酵素の発現が上昇していたのだが (Figure 3A)、実際に乳酸からの糖新生が亢進しているかどうかを確認するために、3つある乳酸の炭素原子の全てを、広く自然界に存在している12Cではなく安定同位体13Cに置換した乳酸をマウスに腹腔内投与し、13C標識グルコースを質量分析器で定量することで乳酸由来の糖新生を評価することとした。肝特異的Phd2欠損マウスでは、投与した13C標識乳酸から糖新生によって新たに合成されたグルコースが対照群より有意に多いことが確認できたため、肝臓においてPHD2を阻害して低酸素応答を活性化すると乳酸からの糖新生が亢進することが確認できた (Figure 3B)。
また、致死量の乳酸を腹腔内投与して作製した乳酸アシドーシスモデルにおいても、肝特異的Phd2欠損マウスでは血中乳酸値が速やかに減少してマウスの生存率が対照群よりも劇的に改善していたことから、肝臓でPHD2を抑制して低酸素応答をONにすると、肝における乳酸からの糖新生が活性化し、血中乳酸値を低下させ、乳酸アシドーシスの生存率を改善できることが明らかとなった (Figure 3C)[5]。
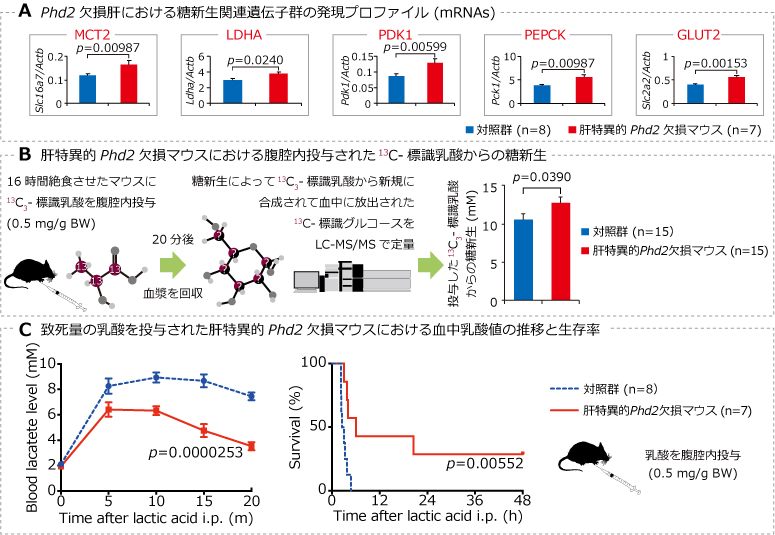 Figure 3. 肝での低酸素応答による乳酸からの糖新生の活性化
Figure 3. 肝での低酸素応答による乳酸からの糖新生の活性化
(A) Phd2 を欠損した肝臓では乳酸からの糖新生に関与する遺伝子群の発現が上昇している。肝特異的Phd2欠損マウスでは、体外から投与した13C標識乳酸からの糖新生が有意に亢進しており(B)、その結果として血中乳酸値が速やかに低下してマウスの生存率も改善することが明らかとなった(C)(文献5より改変)。
【PHDを標的としたメトホルミン関連乳酸アシドーシス (MALA)治療法の開発】
乳酸アシドーシスは非常に重篤な病態ではあるが、なんの原疾患もなくいきなり発症することは、ない。敗血症などの重症感染症、臓器虚血、ミトコンドリア病など、原因となる病態はいくつも知られているが[6-8]、そのなかで、筆者らは全世界で1億2千万人以上が服用しているⅡ型糖尿病治療薬メトホルミン[9]によって誘発される乳酸アシドーシス (MALA: metformin-associated lactic acidosis)に着目した[10, 11]。
MALAは発症率こそ低いが、ひとたび発症すると致死率が50%を超える非常に危険な病態である[10, 11]。特に(高齢者を含む)腎機能低下症例へのメトホルミン投与は、MALA発症のリスク・ファクターとなる。
先述したとおり肝臓でPHD2を阻害することで血中乳酸値を低下できる[5]のであれば、メトホルミン内服などによって発症した乳酸アシドーシス MALAをPHD2の阻害剤で治療できないかと我々は考えた。そこで、0.2%アデニン含有食を給餌して作製した腎機能低下マウス[12] (Figure 4A)にメトホルミンを内服させることでマウスMALAモデルを作製し (Figure 4B-C) 、血中乳酸値が8 mMを越えた個体に対し、vehicleあるいはPHD阻害剤によって治療するモデルを作製した (Figure 4D)。経口投与された薬剤は消化管から吸収されたあと先ず肝臓へと運ばれることを考慮し、PHD阻害剤の投与ルートとして経口投与を選択した。
結果は予想通りであり、PHD阻害剤で治療した群はvehicle治療群と比べて劇的に生存率が改善され、PHD阻害剤がMALAの治療薬として有用であることが確認できた (Figure 4E)[13]。
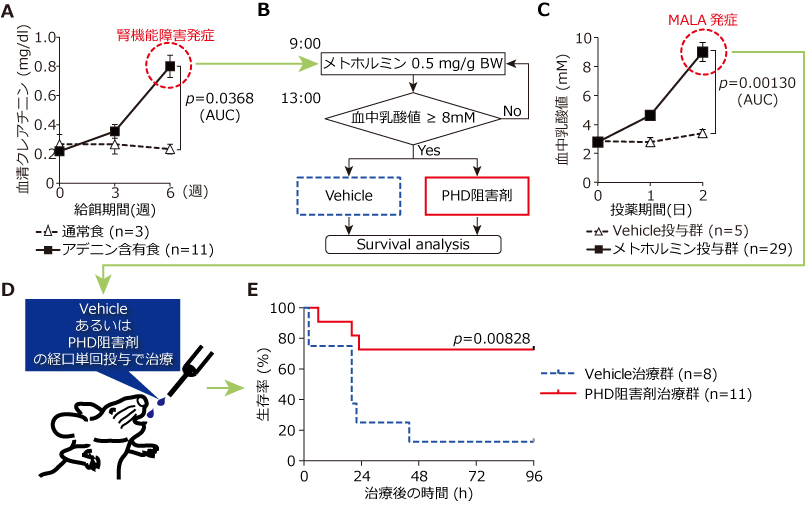
Figure 4. PHDを標的としたメトホルミン関連乳酸アシドーシス (MALA)治療法の開発
(A) 0.2%アデニン含有食を給餌されたC57BL/6マウスにおいては、6週目には血清クレアチニン値が上昇し腎機能障害が誘発されていることがわかる。その腎機能障害マウスに対してメトホルミンを投与してメトホルミン関連乳酸アシドーシス (MALA)を発症させ、PHD阻害剤あるいはvehicleの単回経口投与によって治療を行う(B-D)。PHD阻害剤投与群においてはMALAの生存率が劇的に改善することがわかった(E)(文献13より改変)。
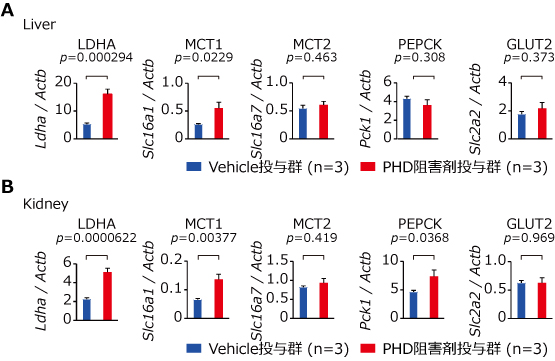
Figure 5. PHD阻害剤による肝・腎での糖新生関連遺伝子群の発現上昇
(A) 肝においてはLDHAやMCT1、(B) 腎においてはPEPCKといった、糖新生関連酵素の発現が、PHD阻害剤によって誘導されていることがわかる(文献13より改変)。
【おわりに】
メトホルミンの作用としては、本来の空腹時血糖降下作用以外にも抗老化や寿命延長[14]・発癌予防[15-19]・抗癌作用[9, 20]などが注目されている。また、米国FDAがメトホルミンの投与制限となっていた腎機能の基準をやや緩和させた (https://www.fda.gov/Drugs/DrugSafety/ucm493244.htm)こともあり、今後メトホルミンの内服者数の増加が見込まれ、それに伴いMALA発症件数が今後増加することが懸念される。
PHD阻害剤の単回経口投与によって乳酸からのブドウ糖の合成(糖新生)に関与する遺伝子群の発現が肝臓だけではなく腎臓においても上昇していたことから (Figure 5)、おそらくPHD阻害剤による治療は血中乳酸の肝臓や腎臓への取り込みおよび乳酸からの糖新生を亢進させることで血中乳酸値を低下させ、その結果乳酸アシドーシスの生存率を劇的に改善できるものと考えられた。本研究成果は、MALAだけに限らずこれまで対症療法しか治療法がなかった乳酸アシドーシス全般に対してPHD阻害剤が特効薬となり得る可能性を示している。現在、PHD阻害剤は腎性貧血の治療薬として臨床治験中であるが[21]、今後は乳酸アシドーシスの特効薬としてのリポジショニングが期待されるところである (Figure 6)。
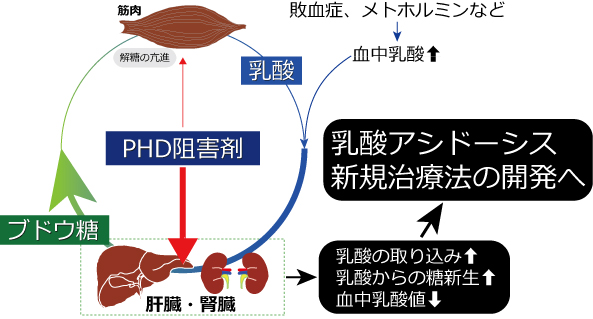
Figure 6. PHD阻害剤による乳酸アシドーシス治療法の概略
PHD阻害剤は骨格筋などの全身の細胞に対しては解糖の亢進によって乳酸産生を促進させるであろうが、肝・腎に対しては乳酸の肝・腎への取り込みと、取り込んだ乳酸からの糖新生を亢進させることで、血中乳酸値を低下させることができるものと考えられる。
【謝辞】
一緒に研究を進めてくださった壽原朋宏先生・小柳津-寅丸智子先生をはじめ慶應義塾大学医学部医化学教室・麻酔学教室・TRセンターのみなさん、PHD阻害剤を御供与くださった第一三共株式会社のみなさん、日々深いディスカッションを重ねてくださっている九州大学生体防御医学研究所中山敬一研・松本雅記研の皆さんに、この場をお借りして深く感謝申し上げます。
【参考文献】
- Kaelin, W.G., Jr. and P.J. Ratcliffe, Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell, 2008. 30(4): p. 393-402.
- Semenza, G.L., Oxygen sensing, homeostasis, and disease. N Engl J Med, 2011. 365(6): p. 537-47.
- Kraut, J.A. and N.E. Madias, Lactic acidosis. N Engl J Med, 2014. 371(24): p. 2309-19.
- Cori, C.F. and G.T. Cori, Glycogen formation in the liver from d- and l-lactic acid. Journal of Biological Chemistry, 1929. 81(2): p. 389-403.
- Suhara, T., et al., Inhibition of the oxygen sensor PHD2 in the liver improves survival in lactic acidosis by activating the Cori cycle. Proc Natl Acad Sci U S A, 2015. 112(37): p. 11642-7.
- Bakker, J. and T.C. Jansen, Don't take vitals, take a lactate. Intensive Care Med, 2007. 33(11): p. 1863-5.
- Angus, D.C. and T. van der Poll, Severe sepsis and septic shock. N Engl J Med, 2013. 369(9): p. 840-51.
- El-Hattab, A.W., et al., MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab, 2015. 116(1-2): p. 4-12.
- Viollet, B., et al., Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond), 2012. 122(6): p. 253-70.
- Inzucchi, S.E., et al., Metformin in patients with type 2 diabetes and kidney disease: a systematic review. JAMA, 2014. 312(24): p. 2668-75.
- Lebacq, E.G. and A. Tirzmalis, Metformin and lactic acidosis. Lancet, 1972. 1(7745): p. 314-5.
- Tanaka, T., et al., Urinary L-type fatty acid-binding protein can reflect renal tubulointerstitial injury. Am J Pathol, 2009. 174(4): p. 1203-11.
- Oyaizu-Toramaru, T., et al., Targeting Oxygen-Sensing Prolyl Hydroxylase for Metformin-Associated Lactic Acidosis Treatment. Mol Cell Biol, 2017. 37(16).
- Check Hayden, E., Anti-ageing pill pushed as bona fide drug. Nature, 2015. 522(7556): p. 265-6.
- Decensi, A., et al., Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila), 2010. 3(11): p. 1451-61.
- Evans, J.M., et al., Metformin and reduced risk of cancer in diabetic patients. BMJ, 2005. 330(7503): p. 1304-5.
- Noto, H., et al., Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One, 2012. 7(3): p. e33411.
- Quinn, B.J., et al., Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab, 2013. 24(9): p. 469-80.
- Zhang, Z.J., et al., Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: a meta-analysis. Diabetes Care, 2011. 34(10): p. 2323-8.
- Wheaton, W.W., et al., Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife, 2014. 3: p. e02242.
- Minamishima, Y.A. and W.G. Kaelin, Reactivation of Hepatic EPO Synthesis in Mice After PHD Loss. Science, 2010. 329(5990): p. 407-407.

