





ホットトピックス
オートファジーと肝疾患
小松雅明
東京都医学総合研究所 蛋白質リサイクルプロジェクト プロジェクトリーダー
サマリー
オートファジーは莫大な量の細胞質成分を取り囲んだオートファゴソームがリソソームと融合することにより執行される大規模な分解経路である。1990年代の出芽酵母におけるオートファジー必須遺伝子ATG(autophagy-related genes)の同定以降、さまざまなモデル生物において遺伝学的解析が行なわれ、現在、オートファジーは飢餓応答だけでなく細胞内恒常性維持、自然免疫、獲得免疫、神経変性疾患抑制そして腫瘍抑制まで関与することが明らかになった。本稿では、肝臓特異的オートファジー欠損マウスから判明したオートファジーの生理機能とその破綻による病態を中心に概説する。
はじめに
細胞内には大きく分けて2つのタンパク質分解経路、すなわちユビキチン–プロテアソームシステムによる選択的タンパク質分解とオートファジー–リソソームシステムによるバルクなタンパク質分解があり、この2つの経路が独立に、時には協調的に働くことにより細胞の恒常性が維持される。オートファジーは細胞外環境に応答して出現した隔離膜が伸長して細胞質成分をランダムに取り囲んだ脂質二重膜構造体(オートファゴソーム)が形成される過程と、生じたオートファゴソームにリソソームが融合して内容物を消化する過程から構成されている。このオートファジー–リソソーム系は、オートファゴソーム内にトラップされたタンパク質をアミノ酸にまで分解することができる基本的に非選択的な大規模分解系であり、新しい膜形成と連動している巧妙かつ複雑な細胞内分解機構である(図1)。この分解系は、栄養飢餓により激しく誘導されることから、自己タンパク質の分解によるアミノ酸供給を介した究極の生存戦略と考えられてきた。しかしながら、最近、高等動物においてオートファジーは飢餓時のみならず、十分に栄養が供給された状態でも恒常的に起こっているらしいことが示唆されてきた。さらに、ある特定のタンパク質がオートファゴソーム膜上に存在するタンパク質と結合することにより選択的にオートファゴソームに取り込まれることが判明した。即ち、オートファジーは飢餓に対応した生存戦略だけでなく、ユビキチン–プロテアソーム系の様にその選択性を介して重要な生理的役割を持つと考えられる。
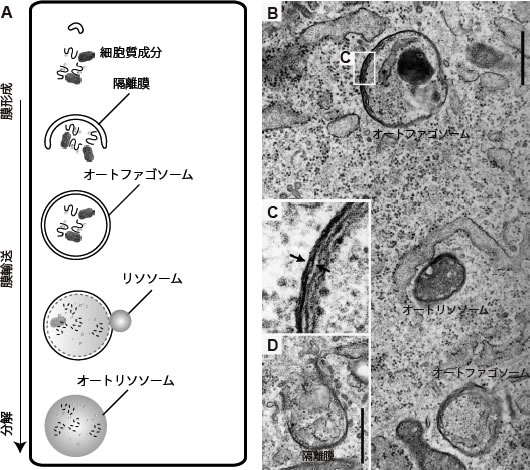
図1 オートファジー。
A. オートファジーの進行過程。細胞内に出現した隔離膜が伸長し、オルガネラを含む細胞質成分を取り囲んだオートファゴソームが形成される。オートファゴソームはリソソームに輸送される。最終的に、リソソームに内在する加水分解酵素がオートファゴソーム内に送り込まれ、内容物の分解が起きる。
B-D. 栄養飢餓後2時間でのマウス繊維芽細胞の電子顕微鏡像。Cはオートファゴソーム二重膜の、Dは隔離膜の強拡大。矢印は、オートファゴソームの内膜および外膜を示す。スケールバー:0.5 um。
1.肝臓のオートファジー
細胞内成分をリソソーム経由で分解する仕組みとしてオートファジーの存在が明らかになったのは、1950年代後半から1960年代前半にかけた頃、即ちde Duveによるリソソームの発見後間もなくである。de Duve、Ashford、Novikoffらは、ラット肝臓の電子顕微鏡観察からオートファゴソームを見出し、オートファジーが栄養飢餓応答として重要であることを最初に示した1)。次いで1975年前後、Mortimoreのグループは、ラット灌流肝臓を用いてこの課題に取り組み、飢餓で誘導されるオートファジーがアミノ酸濃度の低下によること、グルカゴンがインスリンと拮抗してオートファジーを誘導することを明らかにした2)。また同時期、Seglenらのグループは、ラット初代培養肝細胞を用いて特定のアミノ酸除去がオートファジーを誘導すること、また現在も使用されているオートファジー阻害剤3-メチルアデニンを見出した3)。この後、大隅のグループによる酵母オートファジーの発見、そしてAtg遺伝子群の同定により、その分子機構とともに様々なモデル生物におけるオートファジーの生理機能が明らかになり、現在の爆発的な研究展開を迎えている4), 5)。肝臓のオートファジーは、オートファジー研究の先駆けであったわけである。この5年の間に、筆者らが作成した肝臓特異的Atg7(オートファジー必須遺伝子の一つ)欠損マウスから、肝臓オートファジーの病態生理機能が明らかになってきた。
1)飢餓応答
肝臓は、飢餓時に栄養源を供給するため自身のタンパク質を分解することが知られている。事実、野生型マウスでは、48時間の絶食により肝臓の総タンパク質量の30〜40%が消失し、肝臓および血中のアミノ酸濃度が一過的に上昇する。一方、肝臓特異的Atg7欠損マウスでは、絶食に伴う肝タンパク質量の減少が確認されず、アミノ酸の上昇も確認されない。重要なことに、野生型マウスは一過的なアミノ酸上昇以降に血糖値が維持されたが、変異マウスでは低血糖となった。このことは、絶食時における糖新生に肝臓のオートファジーが重要な役割を担うことを示唆している6)。
2)恒常性維持
高等動物においては、栄養が豊富な状態においてもオートファジーは基底レベルで起こっている。実際、初代培養肝細胞などでは富栄養下でもオートファゴソーム形成も、長寿命タンパク質の分解も確認される。条件付きAtg7 ノックアウトマウスを用い1ヶ月間肝臓においてAtg7を欠損させたマウスは、肝実質細胞の膨張や一部に肝細胞死を伴った重篤な肝肥大、肝障害を引き起こす(図2A)。超微細形態解析から、その肝実質細胞内には、変形したミトコンドリア、ペルオキシソーム、小胞体膜が渦をまいたconcentric membranous structuresが蓄積しており(図2B)、オートファジーによる選択的な変性オルガネラ分解機構を支持する。さらに、驚いたことに、変異肝実質細胞において多数のユビキチン陽性のタンパク質凝集体が観察された。これらのことは、栄養条件に係らず起こっているオートファジーが細胞内成分を代謝することにより、常に細胞内を浄化していることを意味する7)。
3)腫瘍抑制
水島らと筆者らのグループは、肝臓オートファジーの長期的な抑制により腫瘍が形成されることを報告した8), 9)。全身性にオートファジー必須遺伝子Atg5をモザイク状に欠損させたマウスや肝臓特異的Atg7欠損マウスは、7から9ヶ月齢で小さな腫瘍が肝臓で検出される。加齢とともに、腫瘍の数、大きさは増加し、16〜19ヶ月齢には肝臓はほぼ腫瘍で覆われる(図2A)。長期的にオートファジーを欠いた肝実質細胞は機能を消失した異常ミトコンドリアを蓄積しており(図2B)、酸化ストレスを被っていた。その結果、ゲノム不安定性が生じ、腫瘍化すると想定される。また、詳細は触れないがオートファジー選択的基質p62の蓄積によるNFκ-Bシグナルやアポトーシス活性化の異常も腫瘍形成に関与している可能性がある。オートファジー欠損で確認される腫瘍は転移能の無い良性腫瘍(アデノーマ)で留まる。従って、オートファジーには腫瘍抑制効果がある一方、腫瘍の悪性化にはオートファジー活性が必要であることが示唆される。重要なことに、Atg5モザイク欠損マウスにおいて、肝臓以外の臓器は腫瘍が形成されない3)。

図2 肝特異的オートファジーノックアウトマウスの病態。
A. 16ヶ月齢の肝特異的オートファジー欠損マウス(Atg7f/f:Alb-Cre)およびコントロールマウス(Atg7f/f)の肝臓。変異マウスは重篤な肝肥大、そして腫瘍形成を伴う。スケールバー:1 mm。
B. 12ヶ月齢の肝特異的オートファジー欠損マウス(Atg7f/f:Alb-Cre)の肝細胞および腫瘍細胞の電子顕微鏡像。オートファジー欠損肝細胞および腫瘍において、変形したミトコンドリア、ペルオキシソーム、concentric
membranous structuresの蓄積が確認される。アスタリスク:ペルオキシソーム、矢印:concentric
membranous structures、矢頭:変形ミトコンドリア。スケールバー:aおよびc:5 nm、bおよびd:1nm。文献9より改変引用。
おわりに
肝特異的オートファジー欠損マウスの解析から、飢餓応答、恒常性維持、そして腫瘍抑制といったオートファジーの細胞機能が明らかになった。今後はオートファジー誘導剤の開発をはじめとした臨床応用に向けた橋渡し研究が重要であろう。
文献
- De Duve C, Wattiaux R.: Annu Rev Physiol, 28: 435-492, 1966
- Schworer CM, Mortimore GE.: Proc Natl Acad Sci U S A, 76: 3169-3173, 1979
- Seglen PO, Gordon PB.: Proc Natl Acad Sci U S A, 79: 1889-1892, 1982
- Ohsumi Y.: Nat Rev Mol Cell Biol, 2: 211-216, 2001
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y.: Nat Rev Mol Cell Biol, 10: 458-467, 2009
- Ezaki, J. et al.: Autophagy, 7: 1-10, 2011
- Komatsu, M. et al.: J Cell Biol, 169: 425-434, 2005
- Takamura, A. et al.: Genes Dev, 25: 795-800, 2011
- Inami, Y., et al.: J Cell Biol, 193: 275-284, 2011

